Phylogenetic analysis is an efficient approach to select and prioritize antibody candidates. It allows researchers to understand B cell lineages and the affinity maturation process, and expand the candidate pool by combining data from different technologies or timepoints. While algorithms to generate phylogenetic trees have been made available by mostly academic groups, generating comprehensive phylogenetic trees remains challenging for many researchers. In this blog post, we will give a brief overview of how B cell phylogeny differs from species phylogeny, what solutions are out there, and what value this provides to researchers working in the antibody discovery field.
Phylogeny in a nutshell
In evolutionary biology, phylogenetic analysis is widely employed to chart the relation between species, proteins, and genes. How do the genes of a mouse relate to those of a human? Is a mouse more closely related to a human than a chimpanzee? These are the types of questions traditionally answered with phylogenetic analysis. Essentially, it is a generic method to analyze the relationship among various biological entities based upon similarities and differences in the genetic code.
Phylogeny-guided antibody discovery
Similar to species evolution, B cells undergo mutation and selection process that can be studied using phylogenetic trees. The reconstruction of phylogenetic trees of antibody variants has become a common practice in antibody discovery workflows. It facilitates selection of high-potential candidates to further test in the lab. There are several benefits of phylogeny that guide the selection of good antibody therapeutic candidates:
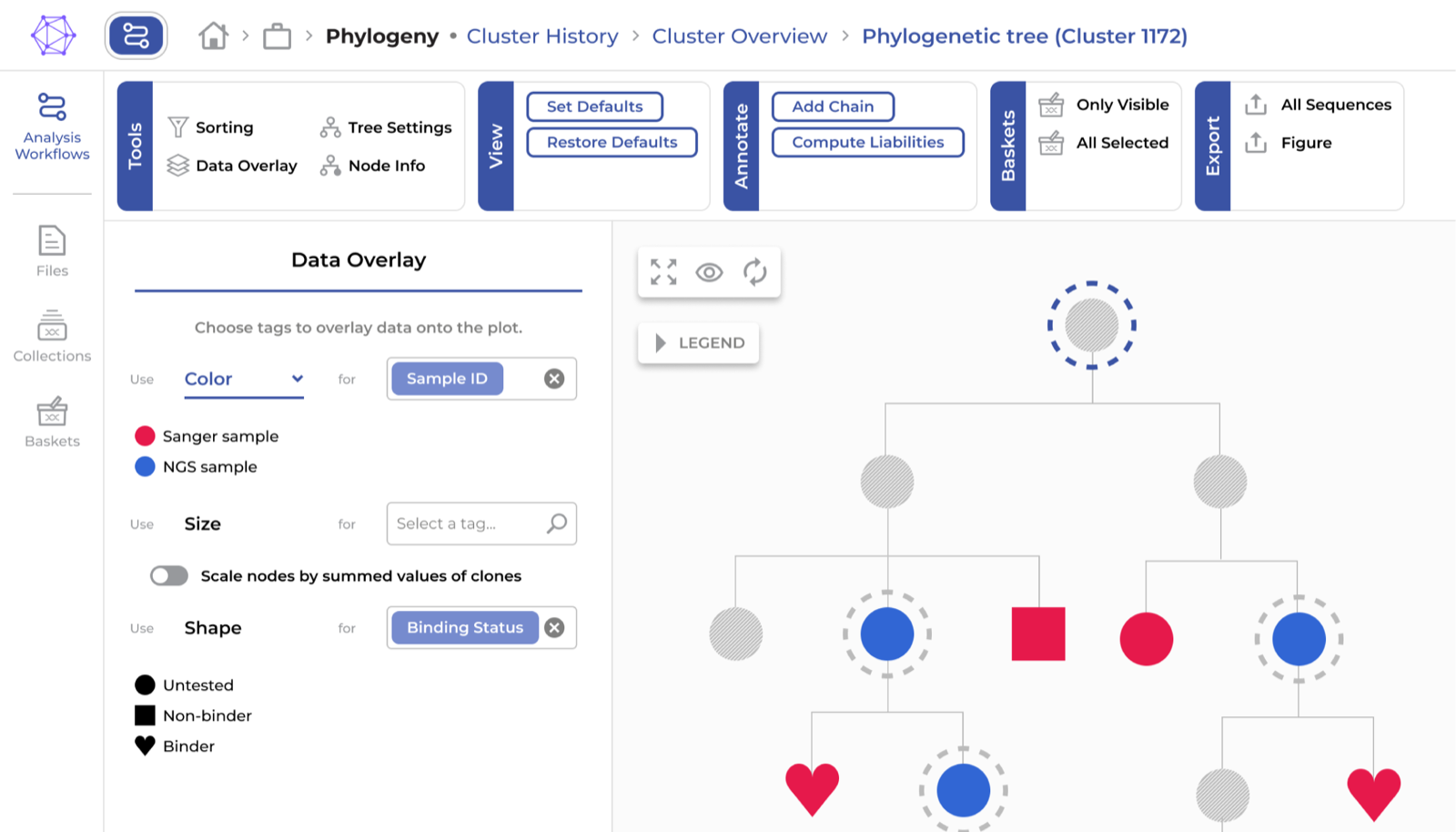
- Clonal expansion – The structure of phylogenetic trees, their size, and node counts can be used to visualize clonal expansion, an aspect that can indicate a response to a target molecule.
- Affinity – The mutational distance of a sequence to its germline can be used to scale branches in the phylogenetic tree. This allows for the identification of heavily mutated variants, which can indicate improved affinity. Alternatively, candidates with readily measured affinity can be used as a visual annotation on the phylogenetic tree to find closely related clones that may have an even larger affinity.
- Affinity over immunizations – In chronic disease or multiple immunizations, B cells might undergo several rounds of affinity maturation. Sampling in-between these instances and plotting phylogenetic trees that contain all samples visually separated by color, helps understanding which mutations were generated in each one of the phases to adjust the binding and could be necessary to capture in a candidate antibody. This is especially interesting in immunizations performed with variants of an antigen where the B cell mutations have to be of a higher degree and end up targeting highly conserved epitopes.
- Sequence liabilities – Annotating the presence or absence of unfavorable characteristics (e.g., hydrophobic regions or exposed cysteines) on the phylogenetic tree can serve as exclusion criteria to narrow down the pool of antibody candidates.
Phylogenetic analysis of B cell repertoires is similar to that of species evolution, but there are notable differences, both in application as well as the associated technical challenges. In order to obtain scientifically robust results, it is important to be aware of these biological differences and to use the right tools for the job.
The peculiarities and pitfalls of B cell phylogeny
B cell phylogeny requires a different technical approach from species phylogeny for several reasons:
- For B cell phylogeny, a constant mutation rate is not assumed, as somatic-hypermutation (SHM) is known to be dependent on the nucleotide context and presence of an antigen. Algorithms developed for species evolution often apply a constant mutation rate and therefore may yield inaccurate results.
- Due to clonal expansion, the exact same receptor sequence can be present in multiple cells. This observed sequence abundance can guide the reconstruction of the tree and should therefore be taken into account. Many phylogenetic tree algorithms fail to utilize this factor which decreases the accuracy of the results.
- Observed sequences are not necessarily placed on the leaf/terminal nodes: A B cell that has divided may yield child cells with a different receptor sequence. At the time of sampling, both child and parent may still exist, and they need to be placed as such, with the parental cell occupying an intermediate (non-terminal) node.
- In sharp contrast with species evolution, where all life on Earth is part of a single phylogenetic tree without a known ancestor, the root of a B cell tree is different for each lineage and it can be inferred: it represents the unmutated germline sequence of a given lineage. Algorithms developed for species evolution often do not allow for the reconstruction of rooted trees or lack the option to select a known root.
Various algorithms have appeared over the years that address one or more of these B cell specific challenges. Unfortunately, the resulting algorithms require complex computations and therefore come with limitations. For example, the Matsen group creates Bayesian models to incorporate uncertainty into the tree inference, with the aim to reflect more accurately important parameters in B cell biology (see no. 2 and 3 in the list above). Unfortunately, calculating such Bayesian statistics requires the computation and evaluation of many trees in parallel and this is therefore computationally very expensive. The Kleinstein group has co-developed IgPhyML, an algorithm that uses realistic codon substitution models based on SHM statistics (see no. 1 in the list above). Also here the accuracy comes at a cost: incorporating a more elaborate substitution model considerably impacts computational costs. These fit-for-purpose phylogeny tools yield more accurate B cell trees but are too slow to construct trees for larger lineages.
In addition to B cell specific tools, there are also well-established general-purpose tools like IQ-Tree, which are fast, easy to use, and highly customizable. IQ-Tree allows users to, for example, define the codon substitution model that is used. Tools’ configurability is crucial, since each researcher may have a different aim and preference on what is the best way to model B cell phylogeny. Whether the increase in accuracy of an algorithm outweighs the computational cost, is dependent on project specific variables such as the research aim, data quantities, computational expertise, and timelines.
Advanced phylogenetic analysis with the ENPICOM Platform
Calculating Bayesian statistics and incorporating elaborate substitution models comes at the cost of computational efficiency. Since users often want to construct trees for many lineages, being able to reconstruct a tree in a reasonable amount of time is a must.
To address the computational challenges, we developed specialized ENPICOM Platform features. Regardless of their bioinformatics expertise, researchers can quickly generate and explore information-rich phylogenetic trees. The robust and fast approach we used will allow us to make adjustments in the future, based on developments in the field.
Sequence liabilities and antibody developability
The ENPICOM Platform allows users to identify sequence liabilities that are exposed on the antibody surface. Internally, a 3D structure of your antibody is generated and every liability (e.g., an unpaired Cystein) that is exposed on the protein surface will contribute to a liability score, which is subsequently added as a clone-level tag.
What’s next?
B cell phylogeny remains a complex issue with rapidly evolving and highly promising analysis methods. We are constantly looking for ways to improve and expand phylogenetic tree reconstruction and address B cell phylogeny challenges.
Would you like to know how you can integrate sequencing and assay data and independently perform advanced clustering and phylogenetic analysis to select the best antibody candidates? Contact our engineers to discuss your use case.


