
Built for scientists
- Analyze data independently — no bioinformatics support required
- Integrate and visualize assay data alongside sequencing data
- Bring predictive model outputs into your clone-selection environment
Biologics-specific pipelines built for scientists

Automated. Reproducible. Tailored.
Pipelines designed by a team with over a decade of biologics domain expertise — not a generic toolkit bent to fit your science.
Accessible to agentic AI: future-proof and ready for the infrastructure pharma is building.
Built around your research, your data, and your teams.
From scoping to production in 4–8 weeks. No 12-month implementation projects.
We maintain, update, and support your pipelines — no operational burden on your team.
We adapt to your ecosystem, not the other way around. Our platform connects to the systems you already use (LIMS, ELNs, data lakes, etc.) while mapping naming conventions, ontologies, and semantics to your internal standards. Data flows stay aligned with those same standards, so everything remains compliant with your governance policies.
Your AI agent can run your pipelines directly, with full biologics context.

For a full security overview, talk to our team.
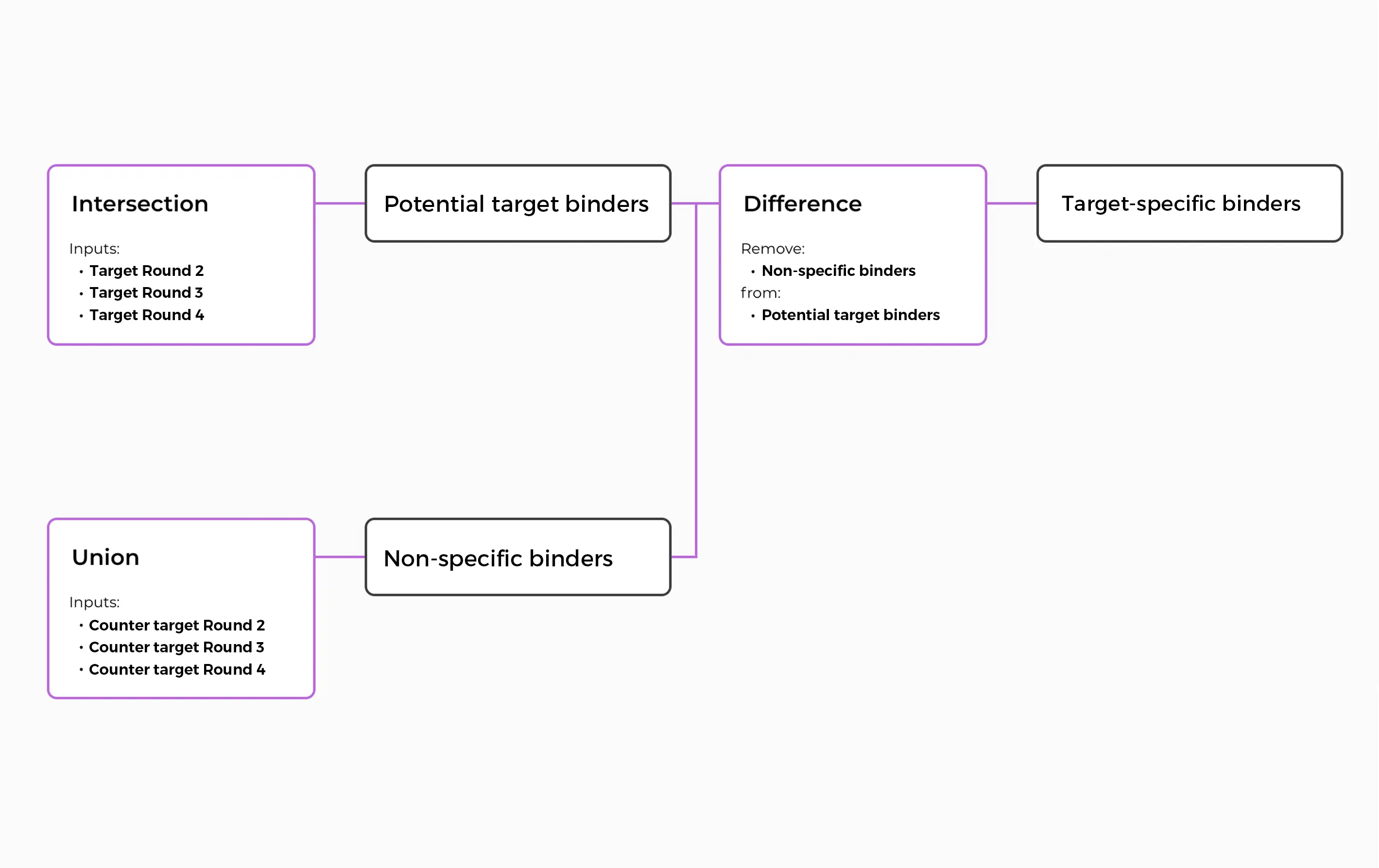
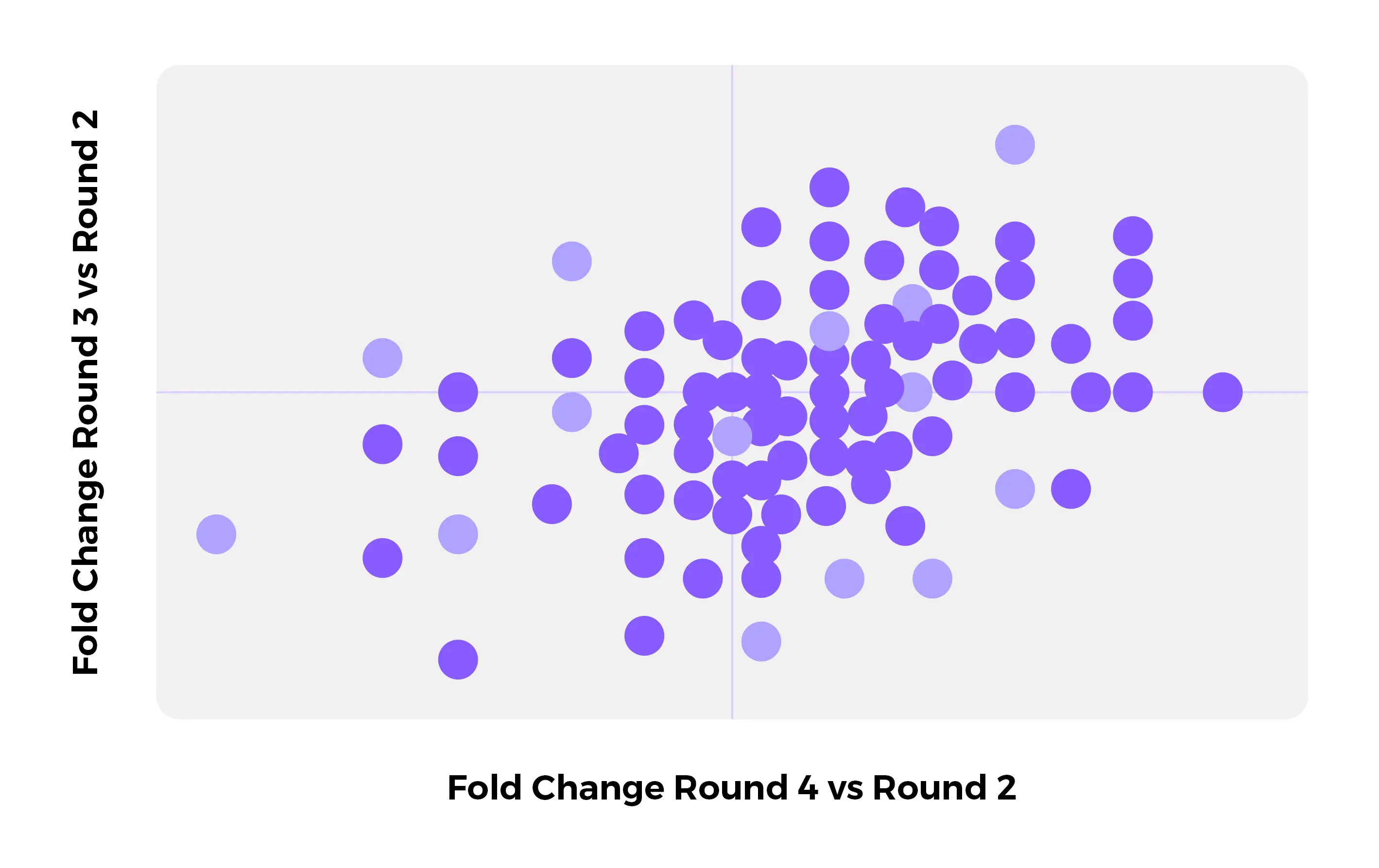
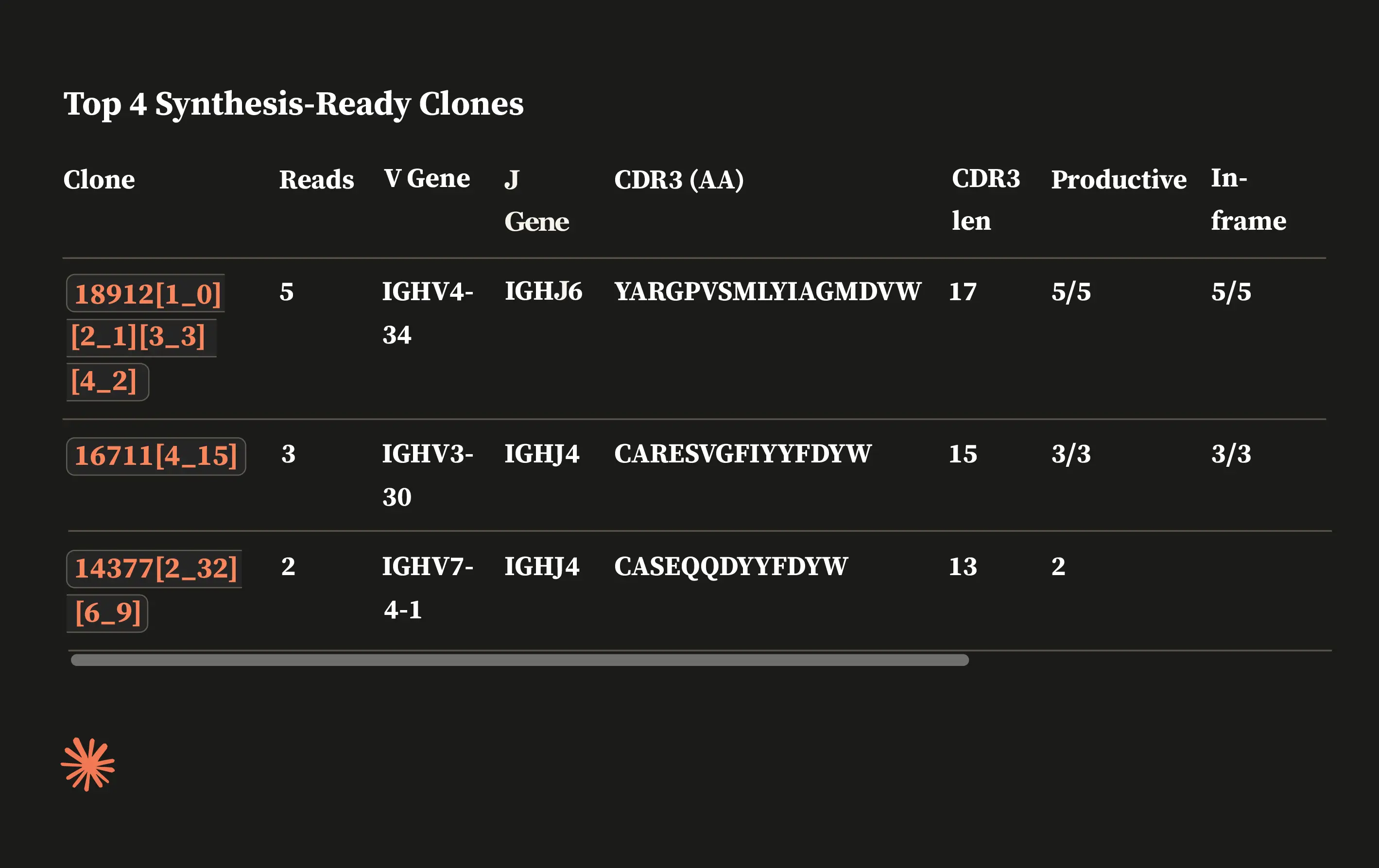
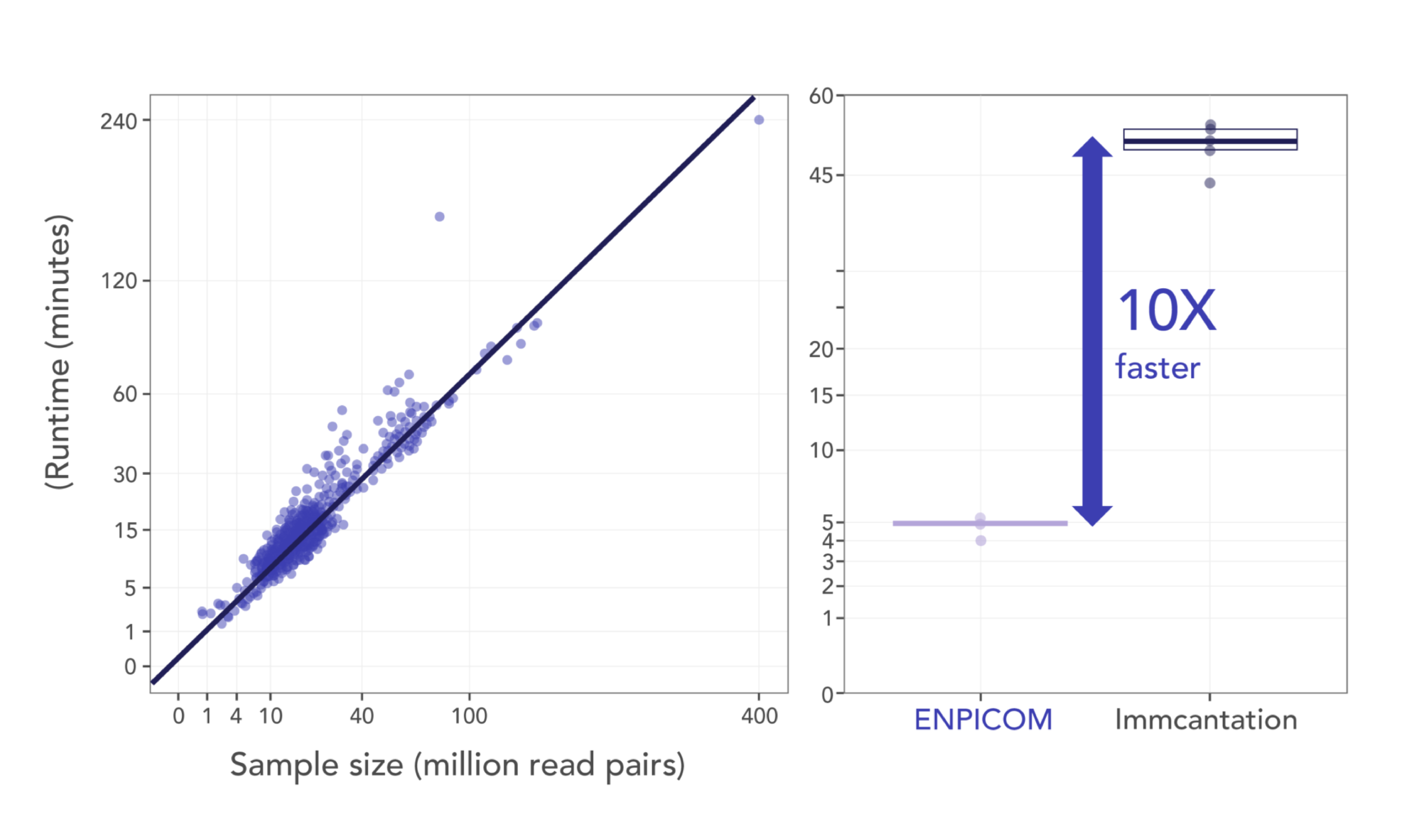
The ENPICOM Platform is a software platform for biologics discovery, best understood as a discovery operating system with an analysis layer on top. It provides an end-to-end environment for analysis-workflow orchestration and covers immune-repertoire and antibody sequence analysis, data integration, assay and screening management, and developability tracking — processing billions of sequences in hours and delivering results directly to scientists, without bioinformatics support.
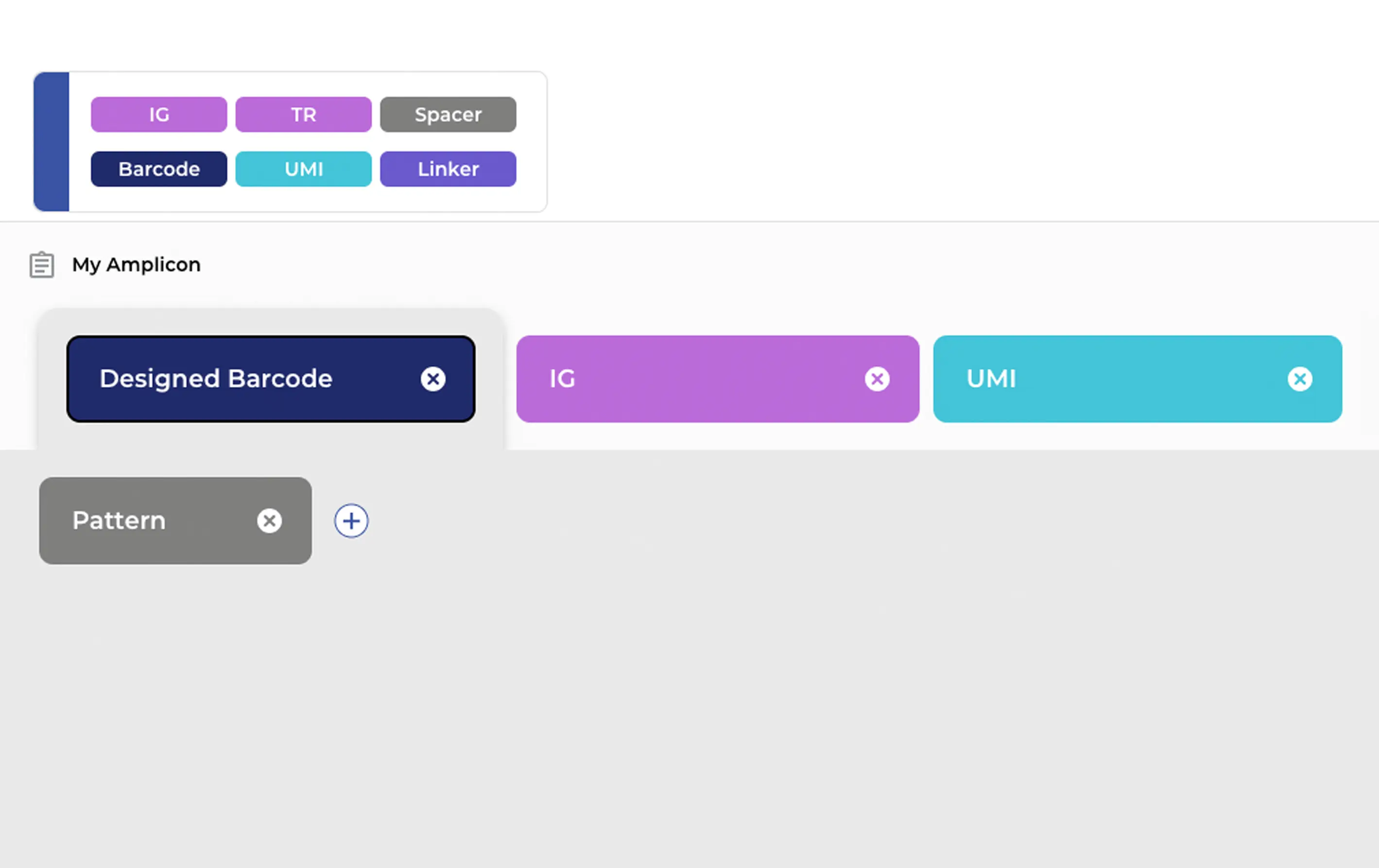
The ENPICOM Platform supports all major biologics sequencing data types: Sanger, NGS, single-cell, and multiomics. It handles any amplicon configuration, including UMIs, barcodes, and linkers, and provides detailed quality-control visualization for both Sanger and NGS data.
The ENPICOM Platform connects to your existing LIMS, ELNs, data lakes, and CRO deliverables. Semantics, ontologies, and data flows are tailored to your internal data-harmonization standards, so the platform fits into your ecosystem rather than forcing migrations, manual transformation, or duplicate data sources.
The ENPICOM Platform exposes its validated pipelines, algorithms, and interactive environments to AI agents through MCP (Model Context Protocol). Agents receive the biologics context they need — mapped to your internal naming conventions and ontologies — so teams can run analyses from their agentic environment alongside the tools they already use.
The ENPICOM Platform is built specifically for biologics, not generic automation adapted for life sciences. Every pipeline is designed around your assay types, sequencing platforms, and SOPs by a team with a decade of biologics expertise, and goes from scoping to production in 4–8 weeks with no internal engineering overhead.
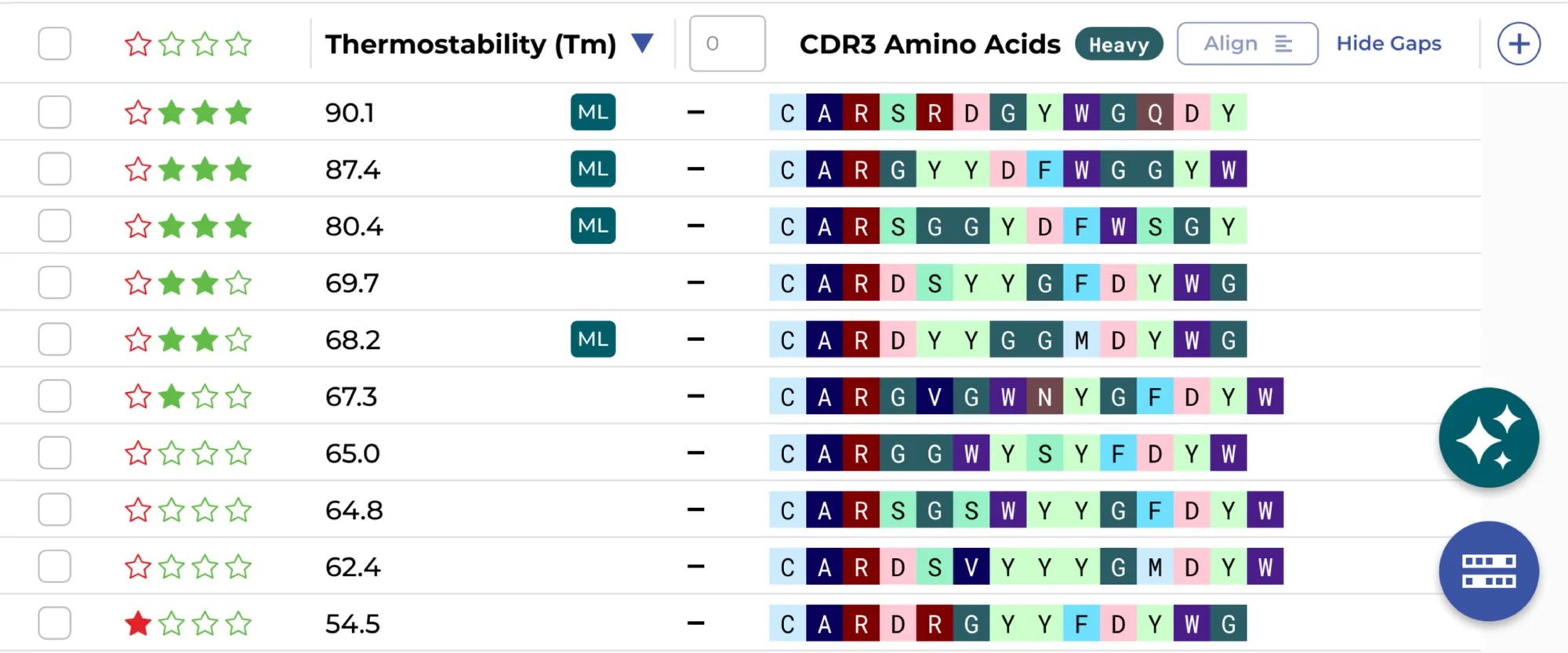
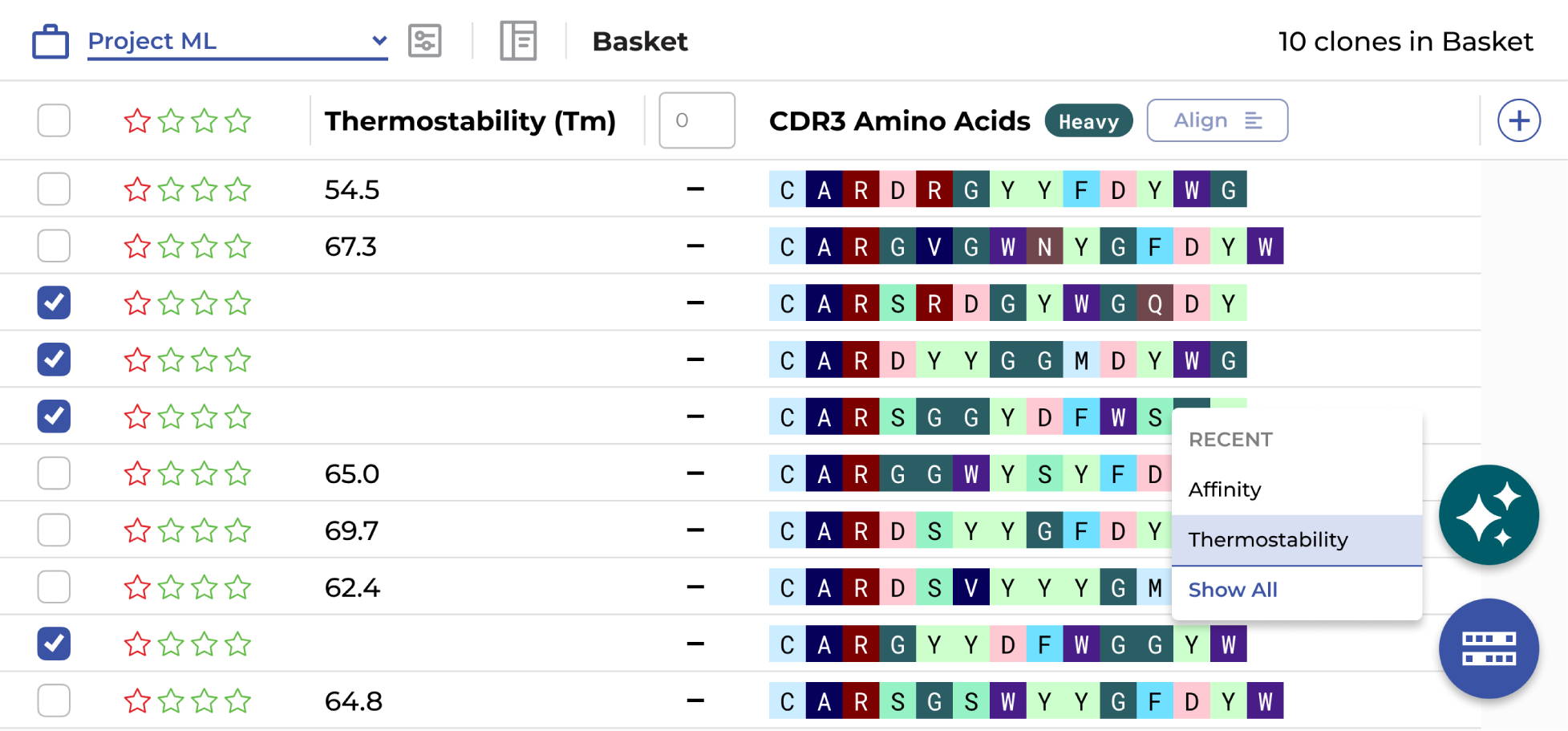
Yes. The ENPICOM Platform is built on open standards with no vendor lock-in. It integrates external and custom ML models directly into the scientific analysis interface — with version control, performance monitoring, and traceability — so data scientists can deploy, compare, and refine models alongside experimental results.
The ENPICOM Platform runs on AWS enterprise infrastructure and is ISO 27001 certified, with single sign-on (SSO), two-factor authentication (2FA), and role-based access control. Reproducibility, audit trails, and governance are built into every pipeline run, backed by regular penetration testing and zero-trust database access.
















