Seamless integration
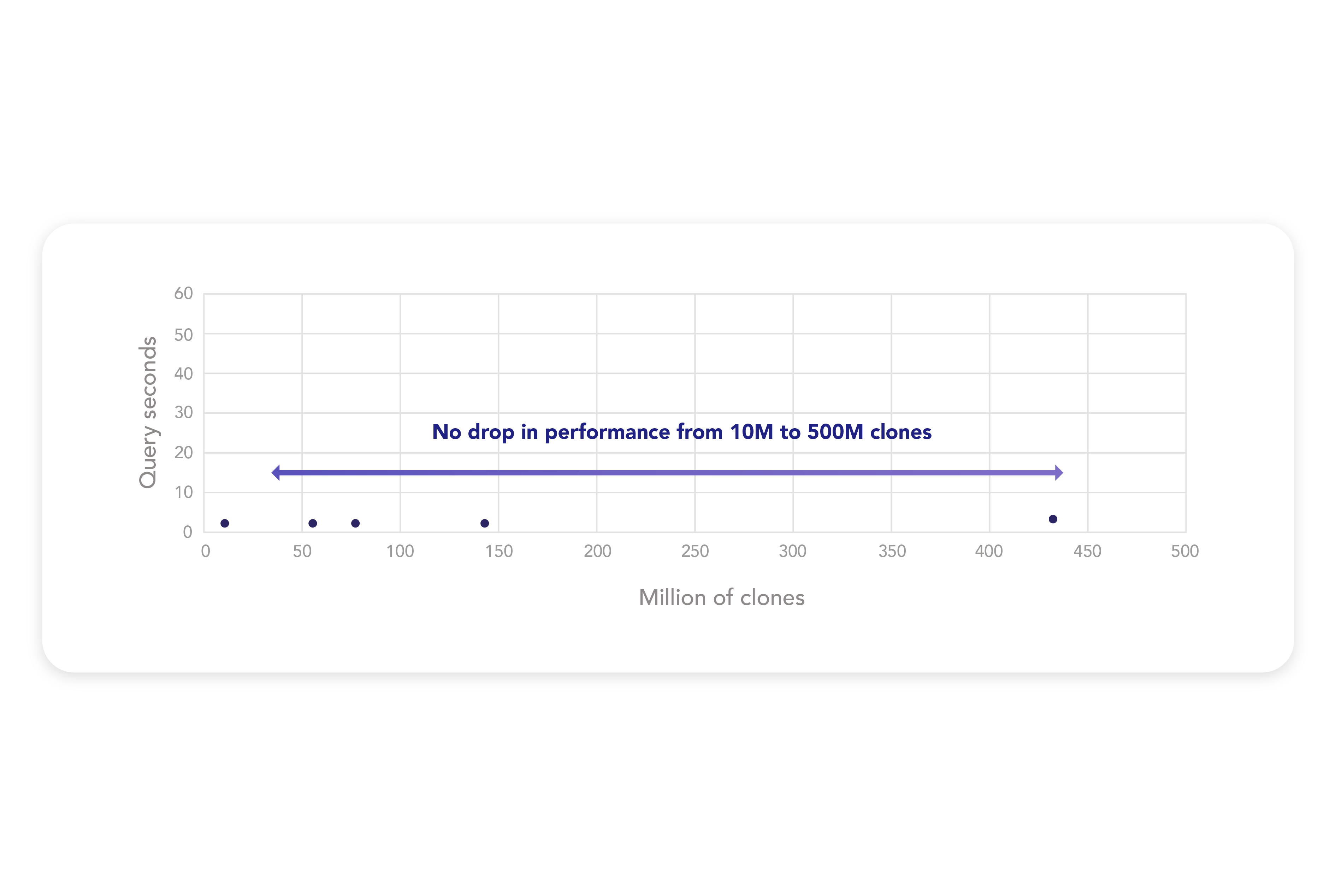
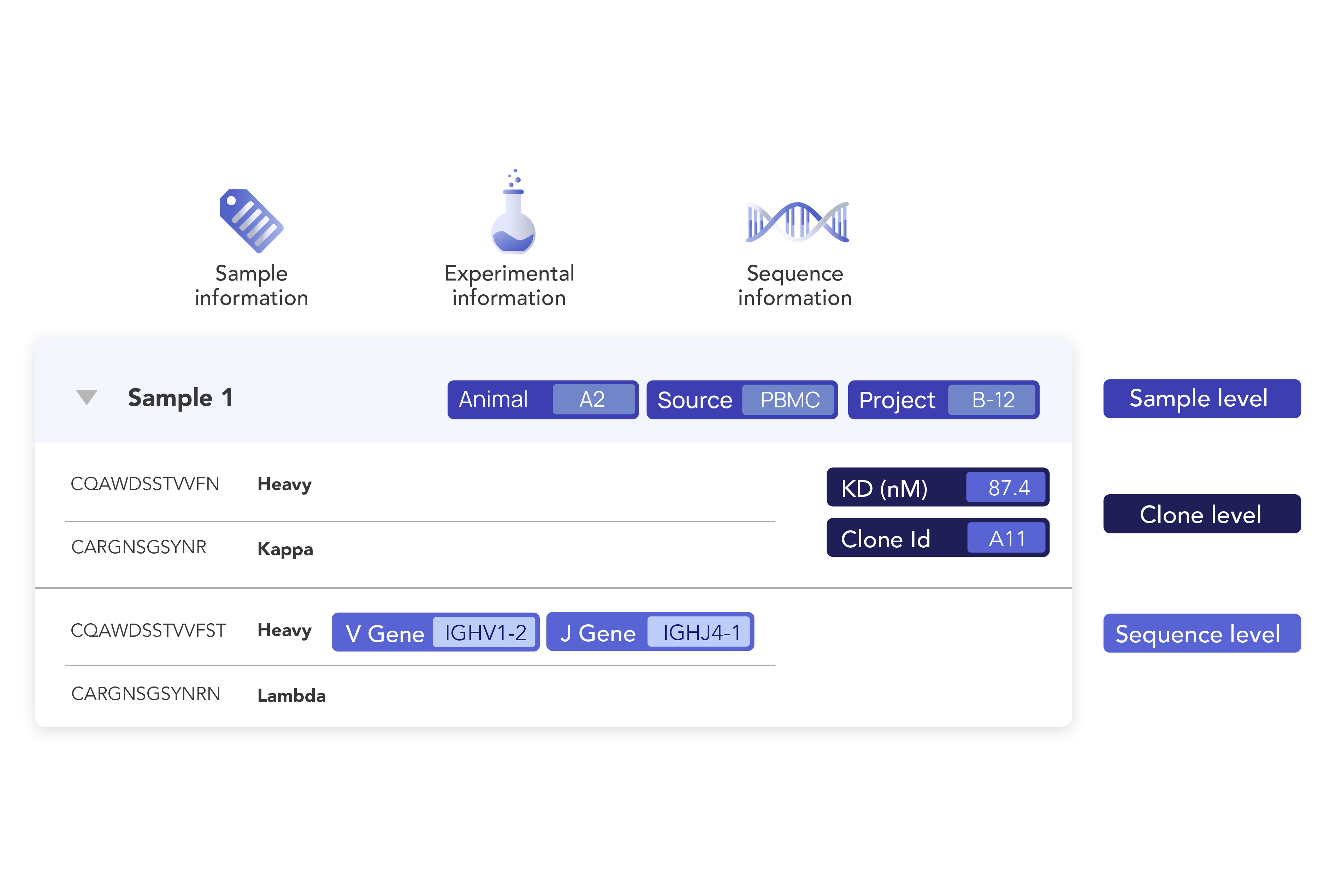
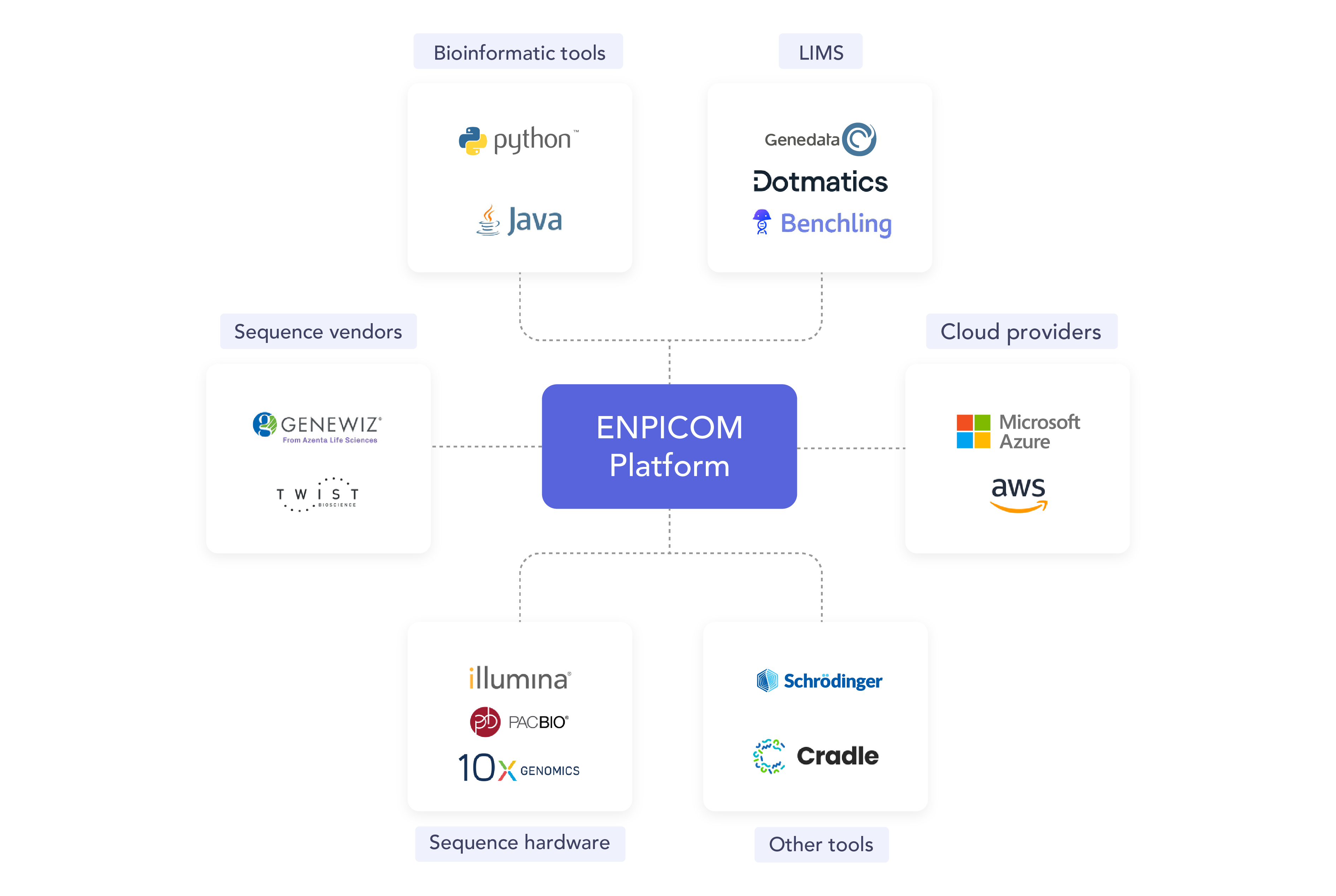
Eliminate data silos and fragmented systems. Our data foundation integrates seamlessly with existing systems and federated data warehouses, creating a single source of truth, FAIR by design, accessible across teams, and optimized for advanced analytics and AI applications.