Recently, the manuscript of the updated read simulation tool InSilicoSeq came out in preprint1 (BioRXiv) and on GitHub (InSilicoSeq 2.0). The main purpose of this package is to generate synthetic datasets that imitate real-world scenarios so that scientists and programmers can validate and optimize bioinformatics tools, statistical algorithms, and analytical pipelines. To extend the InSilicoSeq package from its origin as meta-genome simulator to support the simulation of reads from amplicon-based sequencing protocols, we collaborated with the original package author Hadrien Gourlé. At ENPICOM, we specifically use this package for simulating immune repertoire sequencing (AIRR-seq) data for benchmarking and testing.
In this blog, we would like to showcase how to simulate realistic AIRR sequencing data using InSilicoSeq 2.0 and demonstrate how we use such a golden-standard dataset to test our own IGX Platform. In particular, we are going to test the error correction functionality that uses several reads from the same template to construct a consensus receptor sequencing. To do this, we are going to simulate paired-end reads from immune receptors with a UMI barcode included (Figure 1) using the new amplicon-generation feature that is now included in InSilicoSeq. For every step, we will provide code and an example file so that you can easily reproduce this workflow or adapt it to your own needs.

Figure 1: Template to be constructed for read simulation. It consists of a unique molecular identifier (UMI), a spacer, and an immunoglobulin heavy chain receptor (IGH).
What is InSilicoSeq?
InSilicoSeq is a software package fully written in Python that can simulate realistic Illumina-like sequencing reads for a variety of sequencing machines and assay types. Originally created by H. Gourlé et al.2, version 2.0 now supports amplicon-based sequencing that enables simulation of AIRR sequencing data1. InSilicoSeq comes with pre-made error models of various quality levels for Illumina MiSeq, Hiseq, NovaSeq, and NextSeq sequencing platforms while it also provides the flexibility to generate custom error models. It has improved computational performance with a reduced memory footprint compared to the original implementation. In this blog post, we will demonstrate the novel amplicon-based sequencing capability, how strongly the simulated reads resemble the PHRED scores of actual sequencing data, and how this data can be used to test tools. If you would like to learn more, you can read the manuscript available on the BioRXiv preprint server.
Generating Immunoglobulin heavy (IGH) receptor sequences
First, we generate a set of receptor sequences that will serve as the repertoire that will be used to generate reads. The R-package immuneSim3 published by the Greiff lab provides a good and easy-to-use option to generate realistic Human Immunoglobulin (IG) heavy-chain receptors. ImmuneSIM enables in silico generation of single and paired chain human and mouse B- and T-cell repertoires. The simulation algorithm encompasses an in silico VDJ recombination process with on-the-go detailed annotation of the generated sequences and, if enabled by the user, somatic hypermutation (SHM) and sequence motif implantation.
The panel below demonstrates the R-code used to generate a small set of 1000 Human Ig heavy chain receptor sequences without somatic hypermutations that will serve as the input to generate paired-end reads using InSilicoSeq 2.0. The output of the R-code is provided in the panel below the code. To make the simulated data more realistic, we added a unique molecular identifier (UMI) consisting of 10 random nucleotides followed by a short spacer sequence of “ACTG” to indicate the boundary between the end of the UMI and the start of the receptor sequence.
Creating InSilicoSeq input-files: Fasta library and read-count file
The test amplicon templates are constructed by concatenating the UMI, spacer, and receptor sequences. For each template the value of the column id (an integer ranging from 1 to 1000) is used as the template name. This way, we can always validate the template sequences by checking the receptor sequence in the immuneSim.tsv file. We used the R-package seqinr4 to write the amplicon templates to a fasta-formatted file. The library file contains the final amplicon templates that can be used by InSilicoSeq 2.0 as templates to generate the forward and reverse reads. Here we provide the fasta-file and the R-code to write the fasta file.
Example output file generated by script: hs_igh_naive_01.fasta
Next, we will create the read-count file. The read-count file contains the number of reads to be generated for each fasta template in the library file. This example code generates 10 reads for each of the 1000 amplicon templates in the library file. Using this read-count file produces a simulation with 5000 forward, and 5000 reverse reads, a grand total of 104 reads
Example output file generated by script: hs_igh_naive_01_read_counts.tsv
Installing InSilicoSeq
Now that the required library and read-count files have been generated, InSilicoSeq can be installed to start generating the simulated reads. You can install it directly onto your system via the easy-to-use python3 pip module.
#install InSilicoSeq
pip install InSilicoSeq
# check if installation was succesfulliss generate --helpOr, alternatively you can choose to use the (biocontainer) docker-image of InSilicoSeq:
# pull docker from repo
docker pull quay.io/biocontainers/insilicoseq:2.0.0--pyh7cba7a3_0
# check if docker works
docker runquay.io/biocontainers/insilicoseq:2.0.0--pyh7cba7a3_0 \
iss generate --help Running InSilicoSeq
InSilicoSeq can generate both (meta-)genome and amplicon-based reads. In this example, we use the generate function to simulate reads. We set the --sequence_type parameter to amplicon to simulate amplicon-based sequencing reads. With the --model parameter, we select the error model miseq. Error model miseq is a precomputed error model based on paired-end reads of 301 base pairs generated by an Illumina MiSeq platform (see preprint for more details). For the --genomes parameter we use the previously generated hs_igh_naive_01.fasta file, containing 1000 amplicon templates. We set --readcount_file to hs_igh_naive_01_read_counts.tsv indicating that 10 reads should be generated for each amplicon template. The number of reads to generate is specified per sequence, so any number will do. Since there are error models based on single reads and read-pairs, the counts file contains the total number of reads. In this example we selected the miseq paired-end error model in combination with a total count of 10 reads per amplicon template. This combination of error model and read count file will result in 5 read-pairs. Providing a read count file containing uneven number of reads in combination with a paired-end results in one incomplete pair.
We are using a prefix of hs_igh_naive_01 to name the output meta and fastq files in the current working directory (${PWD}/hs_igh_naive_01). The optional parameter --store_mutations enables that the errors introduced by InSilicoSeq in each read will to be stored as a vcf-formatted file. The --cpus parameter dictates how many CPUs can be used by InSilicoSeq. Putting together these parameters will lead to the following command to simulate reads with InSilicoSeq:
hs_igh_naive_01.fasta and hs_igh_naive_01_read_counts.tsv files.
hs_igh_naive_01_R1.fastq) and reverse reads (hs_igh_naive_01_R2.fastq) each containing 5000 reads. The file hs_igh_naive_01.vcf contains the position of the errors introduced by InSilicoSeq for each individual read. Based on these files, we can further explore the features of the simulated AIRR data.InSilicoSeq produces realistic quality (PHRED) scores
The selection of the error model influences the quality of the simulated data. The FastQC5 tool and fastqcr6 R-package can be used to visualize the average per-base quality PHRED7 scores of the forward and reverse reads generated by the precomputed miseq model. Figure 2 shows the per-base average quality scores of the forward (red line) and reverse reads (green line) of empirical MiSeq data (A) and simulated MiSeq data (B).
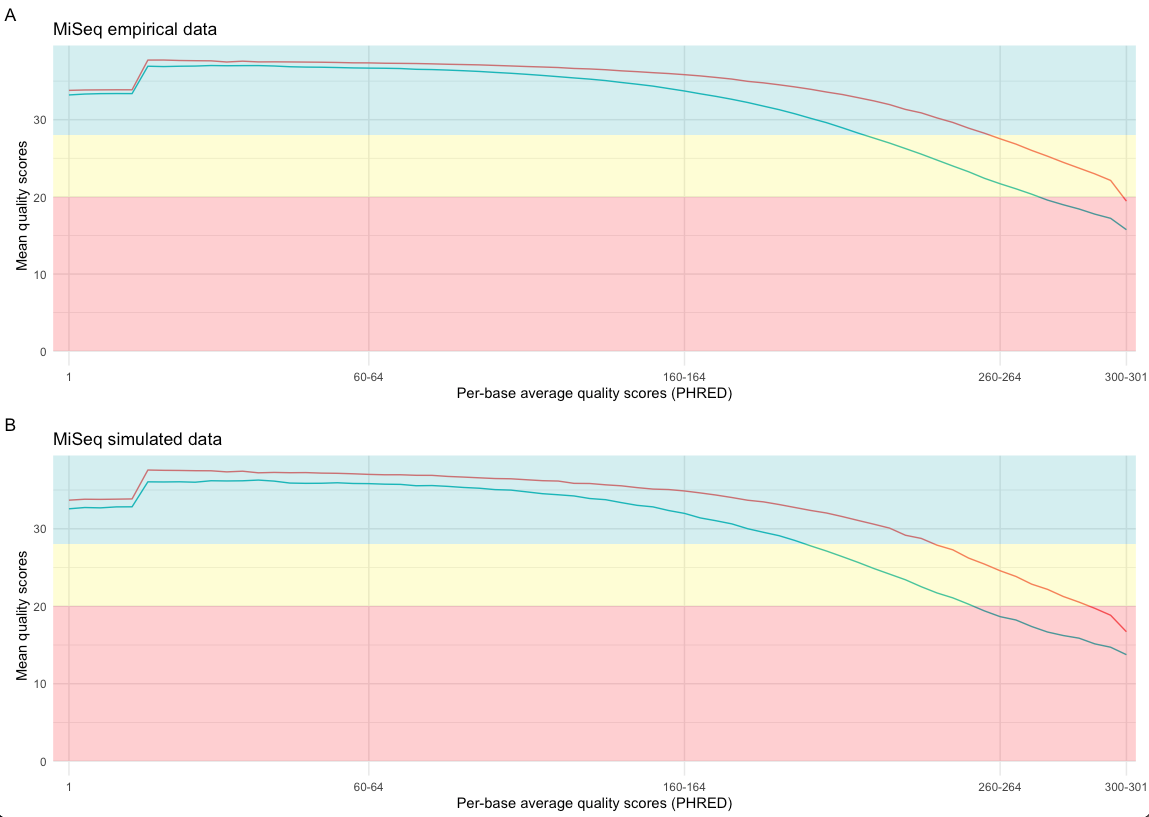
Figure 2: Per-base quality score comparison of empirical and simulated data. The mean quality scores of the forward reads are indicated by a red line and reverse reads by a green line. Subfigure A shows the per-base mean quality scores of empirical MiSeq data and B shows the per-base mean quality scores of simulated MiSeq data. PHRED7 quality scores are analysed by FastQC5 and the figures are generated by the R-package fastqcr6.
Besides the precomputed miseq error model, InSilicoSeq contains precomputed error models from different sequencing platforms and of various quality classes. Table 1 contains the complete overview of the available error models and Figure 3 shows the per-base quality score for each model. InSilicoSeq also has the option (iss model) to create a custom error model based on any BAM file.
Error model | Sequencing platform | Read length | Filters |
NextSeq | NextSeq | 2x 301bp | None |
MiSeq | MiSeq | 2x 301bp | None |
MiSeq-20 | MiSeq | 2x 301bp | Average PHRED score >= 20 |
MiSeq-24 | MiSeq | 2x 301bp | Average PHRED score >= 24 |
MiSeq-28 | MiSeq | 2x 301bp | Average PHRED score >= 28 |
MiSeq-32 | MiSeq | 2x 301bp | Average PHRED score >= 32 |
MiSeq-36 | MiSeq | 2x 301bp | Average PHRED score >= 36 |
HiSeq | HiSeq | 2x125bp | None |
NovaSeq | NovaSeq | 2x150bp | None |
Table 1: Overview of the pre-computed error models for different Illumina sequencing platforms that are available through the InSilicoSeq 2.0 command line interface (cli). For the MiSeq error model, we have created 5 different quality classes by filtering paired reads with different minimal averaged quality scores.
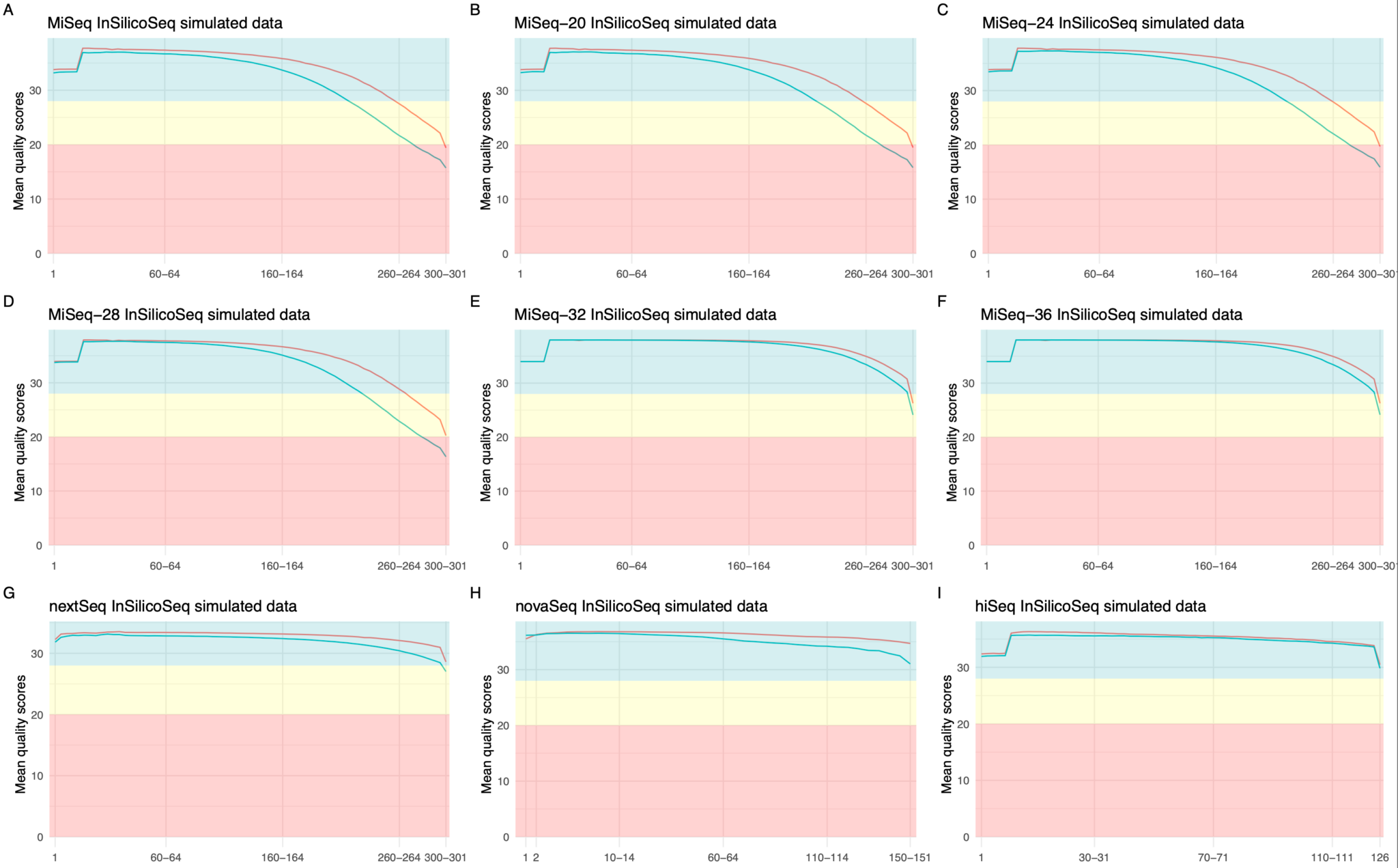
Figure 3: Overview of available error models in InSilicoSeq 2.0. Per-base quality score comparison of empirical and simulated data. The mean quality scores of the forward reads are indicated by a red line and reverse reads by a green line. Each subfigure shows the per-base mean quality scores for the precomputed error models: MiSeq (A), MiSeq-20 (B), MiSeq-24 (C), MiSeq-28 (D), MiSeq-32 (E), MiSeq-36 (F), NextSeq (G), NovaSeq (H) and HiSeq (I). PHRED7 quality scores are analyzed by FastQC5 and the figures are generated by the R-package fastqcr6.
Validating sequencing errors
InSilicoSeq has the option to store the positions of the sequencing errors it simulates on a read. When the parameter --store_mutations is passed, a vcf-formatted file containing the position and error for each read is created. The vcf file can be used as ground-truth data for detecting and correcting sequencing errors and consensus building.
To validate the logging of the sequencing errors, we make a multiple sequence alignment (MSA) of a forward and reverse read to the amplicon template. First, we take the first amplicon template from the hs_igh_naive_01.fasta file and select the first matching forward and reverse read of the two fastq files (hs_igh_naive_01_R1.fastq and hs_igh_naive_01_R2.fastq) (please note that the reverse read must be converted to a forward orientation by taking the reverse complement). This results in the following sequences:
>1is the amplicon template,>1_0_0/1is the first forward-oriented read based of amplicon template>1and>1_0_0/2is the reverse-complemented sequence of the first reverse-oriented read of amplicon template>1. The web-based version of Clustal Omega8 was used to align the two reads to the amplicon template yielding the following result:
Based on the MSA, we can easily identify the position of the errors: A missing asterisk (*) reveals the position of the mismatches between the reads and the template. For read 1_0_0/1 at position 273, an A changed to a T, and at position 299, a C changed to a T. For read 1_0_0/2 at position 70, a G changed to an A; at position 254 an A to a G; at position 258 an A to a G; at position 273, a T to an A and at position 291 an A to a G. The locations of these seven errors could all be validated by the content of the hs_igh_naive_01.vcf file:
This optional vcf-formatted file has the purpose of being used as ground-truth data for detecting and correcting sequencing errors.
Uploading simulated data to the IGX Platform
As we mentioned earlier, simulating reads can be very valuable for benchmarking tool performance. To demonstrate that the sequencing data created by InSilicoSeq can be used for this purpose, we upload the simulated reads into the IGX Platform. The IGX Platform is an end-to-end software solution that we developed to enable any scientist to efficiently analyse immune repertoire sequencing data. Once the read files hs_igh_naive_01_R1.fastq and hs_igh_naive_01_R2.fastq are successfully uploaded into the platform, we need to construct a sequence template.
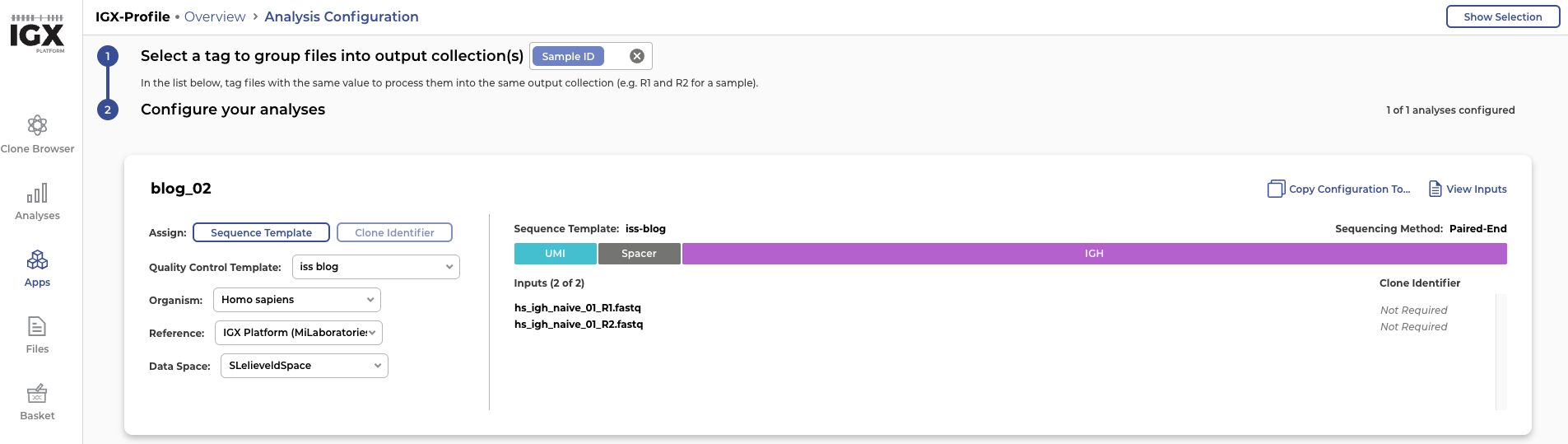
Figure 4: Constructing a sequence template in the IGX Platform: The simulated reads start with a UMI of 10 nucleotides and a spacer consisting of the sequence “ACGT” followed by an immunoglobulin heavy receptor (IGH)
Now we can analyse the simulated reads. For each read pair, we can determine its fate during read processing: For our simulated dataset of 5000 read pairs, around 4% (N=204) were discarded because of quality issues. Based on the detailed read-fate overview, it could be determined that errors altering the spacer sequence and leading to a non-productive CDR3 region are the main reasons for reads-pairs to be discarded. These read fates are expected in our simulated dataset where sequencing errors occasionally alter the spacer sequence or result in a non-productive receptor sequence.
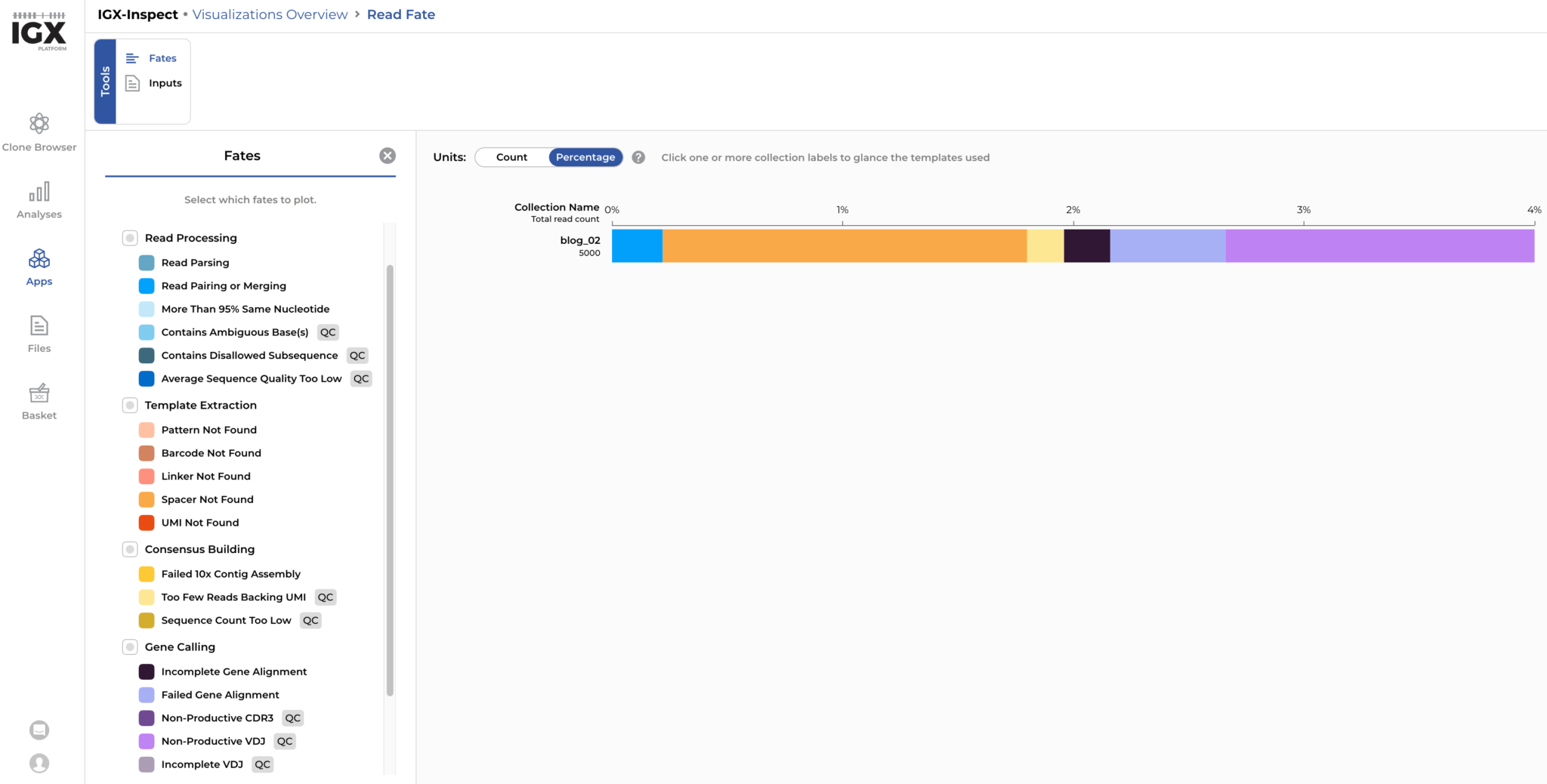
Figure 5: Around 4% of the simulated reads are discarded by the IGX Platform mainly due to sequencing errors altering the spacer sequence and errors leading to non-productive receptor sequences.
Testing the error correction of the IGX Platform using simulated data
The simulated data can serve as a golden standard set to test the error correction and template-building algorithms. In our example, we know the sequence for each amplicon template (hs_igh_naive_01.fasta)
For each amplicon template we know what the simulated forward and reverse reads are based on the fastq read identifiers: The read identifiers of the InSilicoSeq fastq-files are always starting with the header of the template sequence . For this example, we can extract all the reads that are simulated for template >2 (from hs_igh_naive_01.fasta) by extracting the reads that start with @2_ from the fastq files (hs_igh_naive_01_R1.fastq and hs_igh_naive_01_R2.fastq) and convert those to fasta format that will be used later to create an MSA.
For each read, we know the exact position of each error introduced by InSilicoSeq (from hs_igh_naive_01.vcf). Based on the 5 simulated read-pairs and the amplicon template, we can do a MSA to see the distribution of mismatches between the reads and the template using Clustal Omega.
Based on this MSA, we can easily identify the position of the errors: A missing asterisk (*) reveals the position where one of the simulated reads contains a sequencing error. Next, we would like to see if the IGX Platform can reconstruct the template sequence based on the simulated reads that include sequencing errors. We can easily query the output of the IGX Platform analyses on the simulated data and extract the consensus sequence:
To validate that the consensus-building algorithm of the IGX Platform is working correctly, all the read errors should be corrected and the consensus sequence should be identical to that of the amplicon template present in hs_igh_naive_01.fasta excluding the UMI (10 random nucleotides) and the spacer sequence (“ACGT”). Once again, we use Clustal Omega to align the amplicon template sequence >2 of hs_igh_naive_01.fasta to the consensus sequence determined by the IGX Platform. Based on the result, we can see that our software corrected each sequencing error and that the consensus sequence perfectly aligns with the amplicon template that was used to generate the reads.
In this blogpost, we illustrated how we use our newly published tool InSilicoSeq to test IGX Platform, our software solution for immune repertoire data analysis.
- We demonstrated the novel amplicon sequencing feature of InSilicoSeq by simulating a template including a UMI, a spacer and a set of IGH receptors, and generating an AIRR sequencing data consisting of 5000 paired-end reads with a length of 301 base pairs using an Illumina MiSeq-based error model.
- We showed that the InSilicoSeq produces realistic amplicon-based sequencing reads that closely resemble the PHRED scores of empirical reads. Errors are introduced as expected and documented in a VCF file.
- We applied InSilicoSeq’s utility in tool benchmarking by showing that a correct consensus sequence is constructed by IGX-Profile; the simulated read errors are corrected back to the ground truth template that was used for read simulation.
If you are interested in learning how to get the most out of your AIRR-seq data with our tailored software and service solutions, talk to our experts about your research project today.
Authors
 | Dr. Stefan LelieveldBioinformatics Scientist at ENPICOMStefan is a Bioinformatics Scientist holding a Ph.D. in Bioinformatics and Human Genetics. His expertise lies in pioneering the development of cutting-edge algorithms and implementing innovative prototypes to tackle challenges within the immune repertoire sequencing field. | |
 | Dr. Henk-Jan van den HamHead of Research at ENPICOMAs a Head of Research at ENPICOM, Henk-Jan focuses on the latest developments in immunology and bioinformatics, particularly in the analysis of adaptive immune repertoire. With a PhD in Theoretical Immunology & Bioinformatics and post-doc experience in virology, Henk-Jan is responsible for leading academic and industry projects, collaborations, and grant applications in the intersection of immunology, bioinformatics, and software engineering. |
References
- Lelieveld SH, Maas T, Duk TCX, Gourlé H and van den Ham H-J. InSilicoSeq 2.0: Simulating realistic amplicon-based sequence reads. preprint on BioRxiv (2024) doi: 10.1101/2024.02.16.580469
- Gourlé H, Karlsson-Lindsjö O, Hayer J and Bongcam-Rudloff E, Simulating Illumina data with InSilicoSeq. Bioinformatics (2018) doi: 10.1093/bioinformatics/bty630
- Weber CR, Akbar R, Yermanos A, Pavlović M, Snapkov I, Sandve GK, Reddy ST, Greiff V. immuneSIM: tunable multi-feature simulation of B- and T-cell receptor repertoires for immunoinformatics benchmarking. Bioinformatics. (2020) doi: 10.1093/bioinformatics/btaa158
- Charif D, Lobry J. “SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis.” In Bastolla U, Porto M, Roman H, Vendruscolo M (eds.), Structural approaches to sequence evolution: Molecules, networks, populations, series Biological and Medical Physics, Biomedical Engineering, 207-232. Springer Verlag, New York. ISBN : 978-3-540-35305-8. (2007) doi: 10.1093/bioinformatics/bti037
- Andrews S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: Babraham Bioinformatics – FastQC A Quality Control tool for High Throughput Sequence Data (2010)
- Kassambara A. fastqcr: Quality Control of Sequencing Data. R package version 0.1.3, Quality Control of Sequencing Data (2023) CRAN: fastqcr
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Research. (1998) doi: 10.1101/gr.8.3.186
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. (2011) doi: 10.1038/msb.2011.75