Importance of data preprocessing
The sequencing technology used to read these sequences each introduce errors, biases, and artifacts, which need to be identified and corrected. For example, NGS can have quite significant error rates and the library preparation process can lead to amplification biases, especially if multiplex amplification is used [1]. Template switch is not without issues and can lead to longer amplicons and lower quality of merged reads [2]. While NGS has opened the door to the deep interrogation of the immune repertoires, one of the key challenges is still the efficient and accurate analysis of the huge amounts of data that are generated. These datasets are becoming even larger with further advances in sequencing technologies, such as Illumina’s NovaSeq and NextSeq. This makes it increasingly important to have accurate and reliable tools that can scale to extremely large amounts of data.
Due to the diverse and complex nature of immune repertoires, and, moreover, the variety in sequencing protocols, different configurations are required to properly (pre)process the data. Indices, UMIs, and barcodes, when properly integrated, can help remove artifacts, minimize biases, and correct errors during data preprocessing. This is a critical step because the quality of the preprocessed data determines the accuracy and reliability of the subsequent clone annotation step.
IGX-Profile: Accurate clone annotation made easy
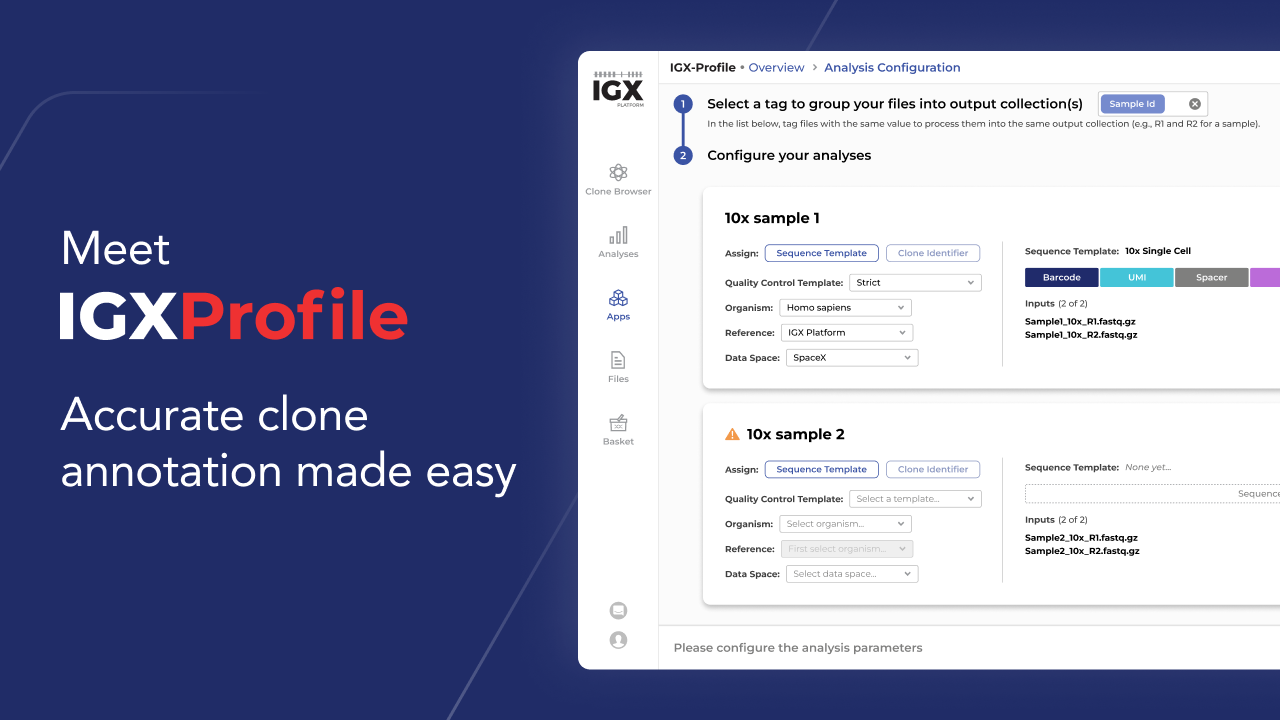
At ENPICOM, we understand these challenges and have developed a solution to address them. Our new immune repertoire processing App, IGX-Profile, is specifically designed to handle any format and amplicon structure, including UMIs, barcodes, spacers, and linkers. It supports error correction and consensus generation, that considers base quality and frequency of occurrence in its proprietary UMI error correction. Extensive QC options allow you to finetune your quality standards and discard low-evidence receptors.
IGX-Profile processes any type of single or paired chain data, using either (1) header or filename information (often used in Sanger sequencing), (2) linkers between chains (e.g. scFv), or (3) barcodes or indices (like 10x Genomics or plate-based single-cell sequencing). Moreover, it is built with a scalability-first approach, processing anywhere from one to one hundred million billion sequences.

Unlock next-generation Rep-Seq data profiling
Whether you are developing new therapeutic antibodies or studying immune responses in the clinic, with IGX-Profile you can be sure that your clones will be accurately annotated and ready to be discovered in one of our downstream analysis Apps.
Would you like to experience IGX Platform’s new data processing capabilities firsthand? Schedule a personalized demo today.
References:
- Barennes, P., Quiniou, V., Shugay, M. et al. Benchmarking of T cell receptor repertoire profiling methods reveals large systematic biases. Nat Biotechnol 39, 236–245 (2021). https://doi.org/10.1038/s41587-020-0656-3
- Vázquez Bernat N, Corcoran M, Hardt U, Kaduk M, Phad GE, Martin M, Karlsson Hedestam GB. High-Quality Library Preparation for NGS-Based Immunoglobulin Germline Gene Inference and Repertoire Expression Analysis. Front Immunol. 2019 Apr 5;10:660. doi: 10.3389/fimmu.2019.00660. PMID: 31024532; PMCID: PMC6459949