News & blog
 White paper
White paperAI in biologics discovery and engineering: A practical guide to driving adoption
Discover how AI can transform biologics discovery and overcome challenges like fragmented workflows, infrastructure limitations, and underutilized models.
 Press release
Press releaseENPICOM joins the Pistoia Alliance
Membership deepens ENPICOM’s commitment to advancing AI and autonomous agent infrastructure for life sciences R&D.
 Webinar
WebinarLibrary to Lead: Rapid In-Lab Sequencing Technologies for Expanding Antibody Discovery
Illumina, Twist Bioscience, and ENPICOM explore an end-to-end workflow for accelerating scFv discovery — from sequencing to synthetic library design to AI-driven analysis.
 Panel discussion
Panel discussionAn honest conversation about what it takes to make ML work in biotherapeutics
Experts from Sanofi, Eli Lilly, Amgen, AbbVie, and Genmab share real-world lessons from adopting AI/ML in biologics discovery and engineering.
 Report
ReportAdopting AI in biologics discovery
Produced with Labiotech: how leading organizations are approaching AI adoption in biologics discovery — what works, what doesn’t, and the capabilities needed to turn AI into measurable impact.
 Blog
BlogUnlocking the potential of AI in biologics discovery
We are at a pivotal moment where AI can transform the future of biologics discovery. By optimizing data for AI-driven workflows, your organization can unlock new possibilities in therapeutic development.
 Webinar panel
Webinar panelBeyond the AI models: What it really takes to deploy AI for drug discovery
A practical discussion on deploying AI for drug discovery, featuring leaders from Genmab, Takeda, and ENPICOM.
 Blog
BlogIntroducing the Models & Molecules podcast
We’re launching Models & Molecules, a new interview series about the innovation at the intersection of biology and computational science.
 Blog
BlogSimulating AIRR-seq runs with InSilicoSeq 2.0
How the new amplicon feature of InSilicoSeq 2.0 lets you generate realistic AIRR sequencing data — and how that golden-standard dataset puts the ENPICOM Platform’s error correction and consensus building to the test.
 Blog
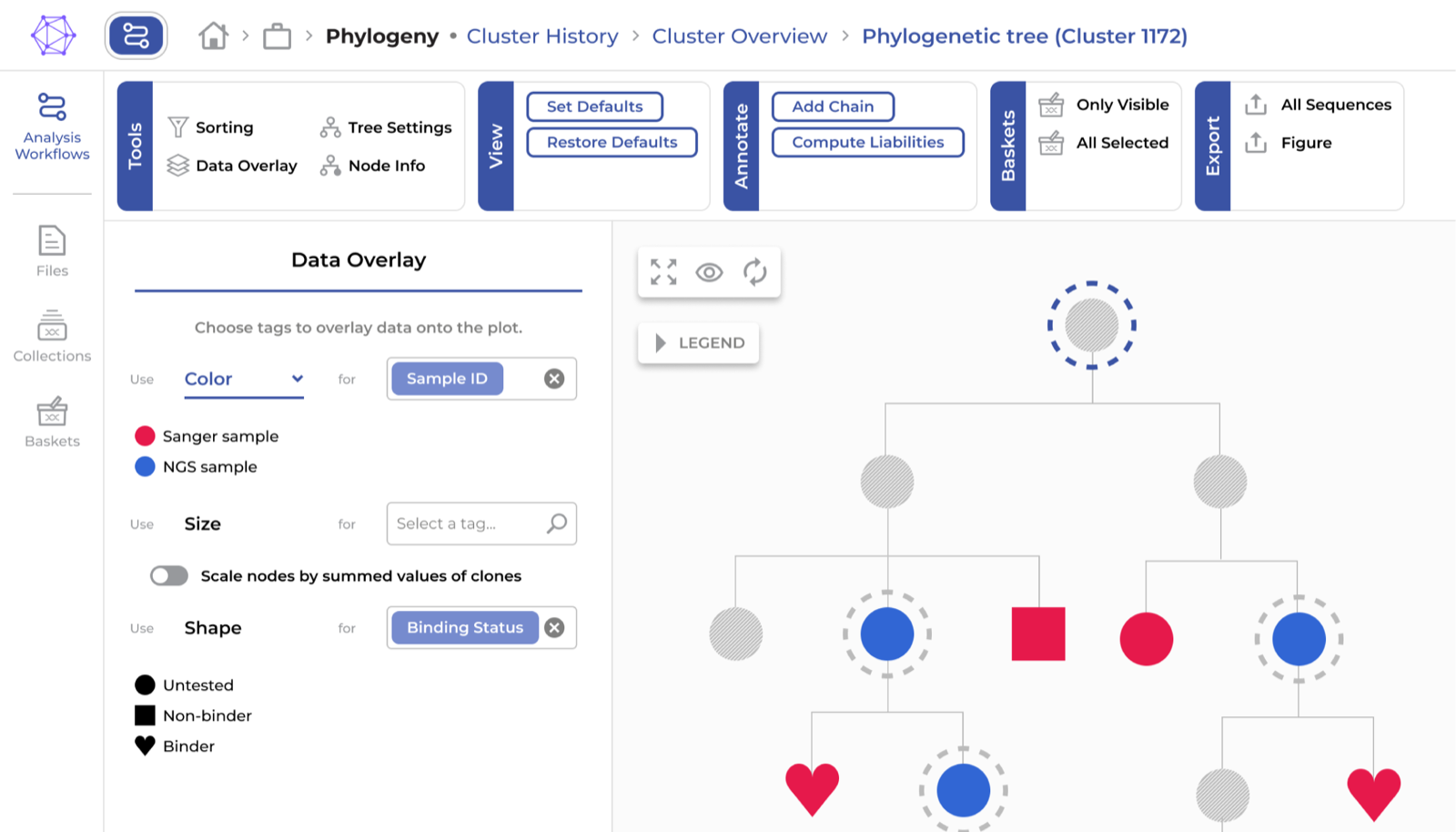
BlogPhylogenetic analysis of BCR repertoires
How B cell phylogeny differs from species phylogeny, the tools available for reconstructing antibody lineage trees, and how the ENPICOM Platform makes advanced phylogenetic analysis fast and accessible.
 Blog
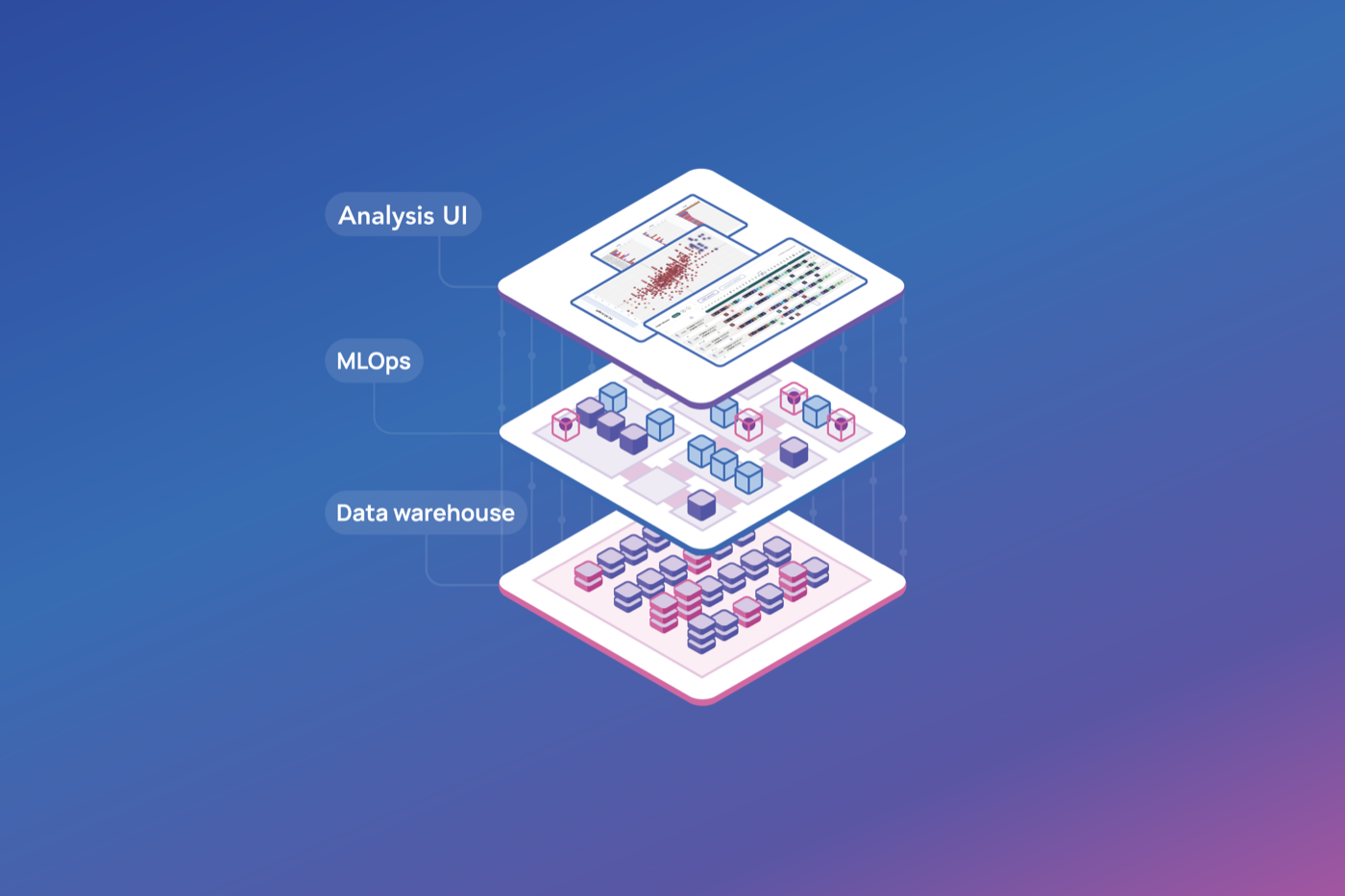
BlogMLOps: The unsung hero of AI-powered biologics discovery
As AI scales across biologics R&D, MLOps — Machine Learning Operations — is what keeps ML models consistent, reproducible, and production-ready. What it is, why it matters, and how ENPICOM connects lab and data scientists.
 Webinar
WebinarDiscovery, characterization & humanization of therapeutic antibodies
A two-part webinar with Genovac, Carterra, and ENPICOM.
 Webinar
WebinarNavigating the immune repertoire
A deep dive into applications, workflows, and new sequencing chemistries.
 Blog
BlogValidating the ENPICOM Platform against a benchmark Rep-Seq dataset
We benchmarked the ENPICOM Platform’s Rep-Seq annotation against a purpose-built public spike-in dataset (Khan et al., 2016) — recovering more receptors and quantifying them more precisely than the study’s original MAF method.
 Blog
BlogAre your scientists making the most of your AI investment?
AI is a powerful tool — but not every researcher is an AI expert. The organizational gaps between your AI investment and real research impact — data, storage, MLOps, inference, and accessibility — and how unified platforms close them.
 Blog
BlogSolving the data connectivity challenge to accelerate drug discovery
Biomedical research generates data at breakneck pace — but it’s only valuable if you can connect it. How APIs bridge siloed data and analysis tools, make legacy data FAIR, and power AI-driven drug discovery.
 Interview
InterviewReducing the barriers for immune repertoire research
A conversation with Dr. Felix Breden on data standardization, sharing, and reproducibility in immune repertoire research — and the ongoing work of reducing the barriers to discovery.
 Blog
BlogEmpowering research teams
Democratizing Rep-Seq data analysis: the challenges of making immune repertoire analysis accessible, the benefits of putting it directly in researchers’ hands, and how to implement a solution that scales.
 Blog
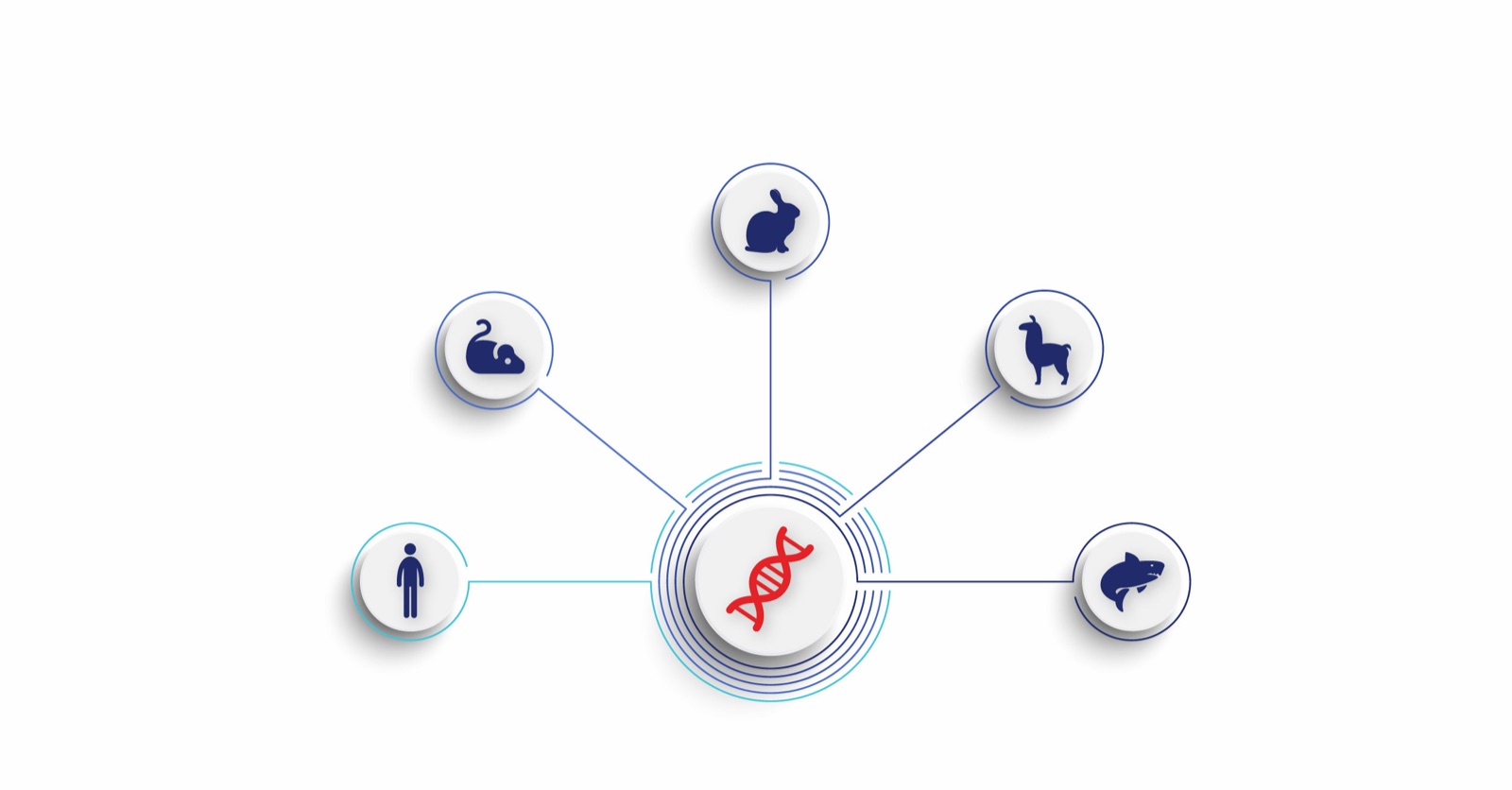
BlogThe animal side of antibody discovery
From mice and rabbits to camelids and nurse sharks — the diverse animal models used in antibody discovery, their roles and trade-offs, and how repertoire sequencing captures the full value of in vivo maturation.