Single-cell sequencing has expanded our toolbox to interrogate the immune system and is increasingly used throughout the entire field of biomedical sciences. In this post, we will take a closer look at the basic principles of single-cell sequencing workflows, discuss data analysis challenges, and explore how Repertoire Sequencing (Rep-Seq) data management is transforming the single-cell data analysis landscape.
Single-cell technology in a nutshell
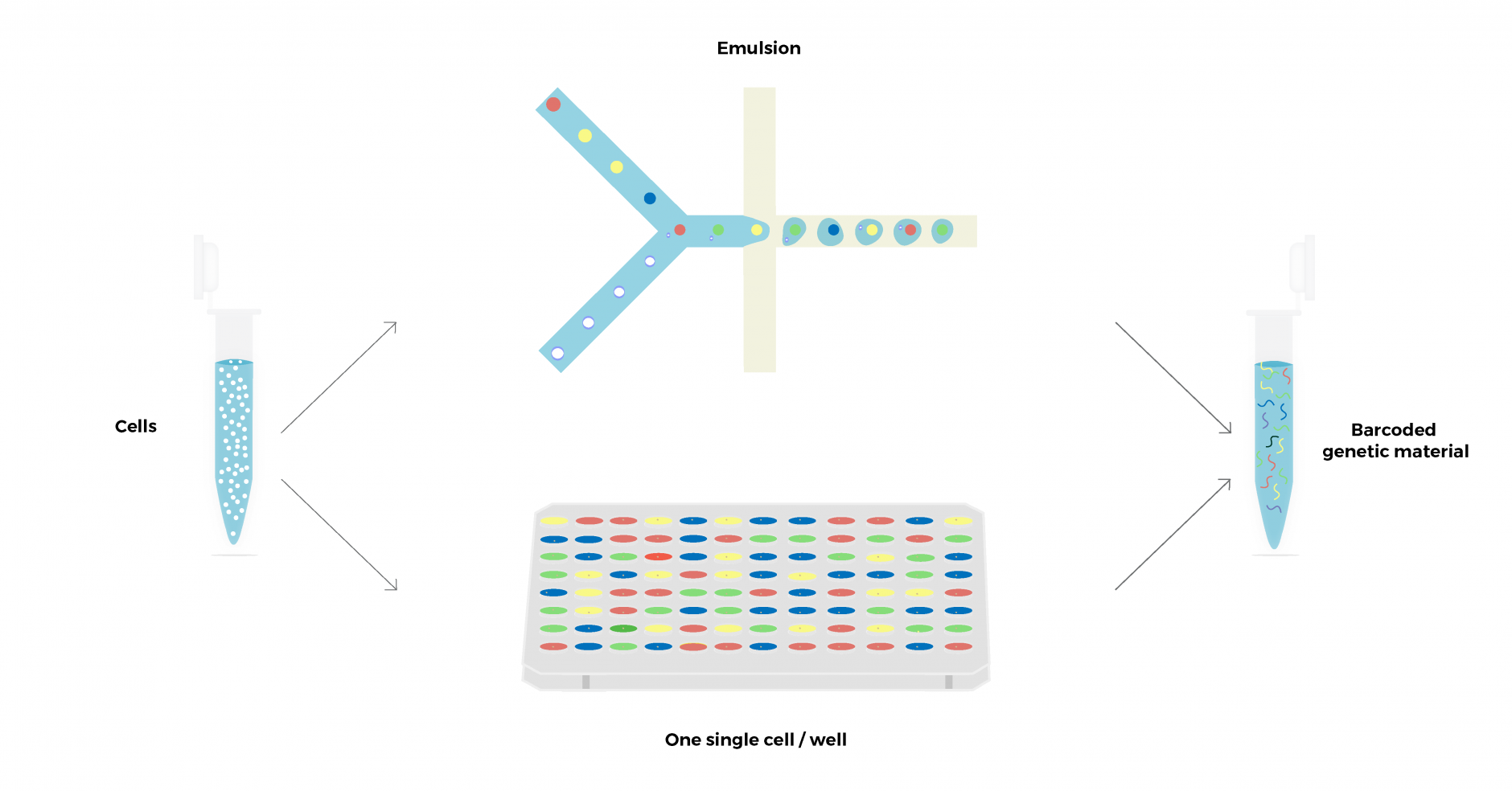
Single-cell sequencing is based on ‘barcoding’ technologies, where unique identifiers are linked to the genetic material obtained from individual cells. The workflow is composed of two main steps: first, single cells are isolated, generally by cell sorting into wells or microfluidic emulsions. Then unique cell identifiers (barcodes) are linked to the genetic material. The barcoded output can be pooled, amplified, and sequenced using standard high-throughput sequencing technologies, allowing for up to thousands of cells to be analyzed in parallel. During the data analysis, the barcode information incorporated during library preparation can be used to identify sequencing reads derived from the same cell.
Being able to capture genetic information from individual cells paves the way for a myriad of novel applications. Here are two of the most promising applications of single-cell sequencing technologies in immunomics research.
Paired-chain information
T- and B-cell receptors (TCRs, BCRs) are composed of two distinct chains, both required to enable antigen binding. Both chains are transcribed from the genome into separate mRNA molecules, which means that traditional bulk sequencing technologies are incapable of capturing the paired-chain information, as there is no way to know the correct pairing of the two chains after the cells have been lysed and sequenced. Conversely, the receptor sequences obtained from a single-cell sequencing workflow can be paired using the cell barcode, providing unprecedented opportunities for fundamental and translational research and drug development.
Multi-omics integration
Barcoded transcripts serve as an ideal starting material for various high-throughput technologies. For example, we can obtain both the receptor sequences as well as transcriptomic data from other genes from the same cell, which can enable the inference of cellular phenotype. Coupling the immune receptor with the cellular phenotype allows us to examine the relationship between the specificity of a cell and its functionality (e.g., cell type, activation status). This greatly improves our ability to understand antigen-specific immune responses and opens avenues for more accurate identification of therapeutic targets, both in terms of finding receptors involved in challenging disease, as well as causative agents.
What are the key data analysis challenges?
The value of any data is determined by the insights and applications derived from its analysis. The interrogation of single-cell Rep-Seq data requires advanced analysis tools as it adds an extra level of complexity to the analysis. Moreover, the integration of single-cell data with bulk repertoire or transcriptomics datasets presents a significant challenge to researchers.
The added layer of data complexity
Compared to bulk repertoire sequencing datasets, single-cell Rep-Seq datasets contain an additional layer of information: the pairing of receptor chains. This extra dimension strongly affects data management and downstream analysis. A clear example that illustrates the added value and complexity of paired-chain information is the grouping of cells into clonotypes. As there is no absolute consensus among immunologists on the definition of a clonotype, for this post we will define it as: “all the cells stemming from a single rearranged progenitor lymphocyte”.
For bulk repertoires, clones are grouped based on a single chain (often the heavy or beta chain for BCRs and TCRs, respectively), but this definition uses only half of the information concerning the cell specificity, thus providing only a partial picture of the clonotype. Paired-chain data contains sequence information of both elements required to reconstruct a functional T or B cell receptor. This more accurate representation of clonotypes, however, makes quantifying cells from single-cell Rep-Seq datasets – with multiple chains – a challenge. Cells can contain multiple receptor sequences, productive and non-productive, leading to complexities when grouping them into clonotypes or comparing them across samples. Most Rep-Seq analysis solutions on the market do not incorporate or support the paired chain information, leading to a loss of valuable (and readily available) information.
The integration with bulk repertoires
Typically, single-cell workflows result in the characterization of a few thousands of cells. This is in sharp contrast with the depth of present-day bulk sequencing experiments, which can contain up to several millions of cells. As a result, single-cell datasets are less suited for analysis of repertoire-wide metrics such as diversity and clonal expansion. By integrating the two technologies, high-resolution, data-dense single-cell datasets can be used to enrich bulk repertoires. This allows both data sources to synergize as they contain complementary information which can be linked. Currently, many available tools are tailored to either the bulk or single-cell data, making the integration and comparison of clones from both technologies a challenging task.
Summing up and a looking ahead
The field of single-cell sequencing is progressing at unprecedented speeds. Continuous technological and scientific advances give rise to the development of novel wet-lab strategies, increased throughput, and improved data quality. In parallel, sequencing costs are decreasing, enabling academic labs and smaller organizations to make single-cell technologies part of their immunomics toolbox.
Single-cell sequencing is revolutionizing the entire field of biomedical sciences. With access to robust, scalable, and intuitive software solutions engineered for the interrogation of these valuable datasets, researchers will be able to fully extract all available insights from the data generated by these technologies.
Learn more about our Rep-Seq data management, analysis, and integration software and how it can advance your immunology research.